

Recently it has come to my attention that there is a strong connection to fibrin, hypercoagulation, biofilms and the embedded infection associated with IC. It is a complex interaction of genetics and environmental influences that leads to this condition. One very strong environmental influence is are infections such as viruses or even their remedies.
The purpose of this blog is to act as a handbook for patients who are trying to understand in more detail various pathways that could be contributing to their health struggles.
Here is a quick summary of the literature.
First a quick summary of the different terms you will see throughout this paper.
Special Thanks to Ruth Kriz for being my mentor and bringing these complex issues to my attention- she has been a wonderful resource for the IC community.
First, let’s go over some of the terminology.
Coagulation definition— the process in which a blood clot is formed. Often referred to as secondary hemostasis.
Components of the coagulation process include:
- Fibrin– stabilizes blood clots and promotes hemostasis.
- Fibrinogen– a glycoprotein found in the blood and is converted to fibrin. It contributes to inflammation, thrombosis, and wound healing (and blood clots)
- Thrombin– also known as factor IIa, enzymatically converts (catalyzes) the conversion of fibrinogen to fibrin.
- Factor V– protein that acts as a cofactor. Activated by thrombin, it can convert prothrombin to thrombin- acting to increase coagulation. Protein C degrades it and stops it activity.
- Plasminogen-activator inhibitor 1 (PAI-1)- an inhibitor of plasminogen- by terminating the activity of tPA- causing a pro-coagulation state. Inhibits fibrinolysis.
- Tissue factor– through the action of cytokines, its activation promotes prothrombin and blood clotting cascade.
- Endotoxin-stimulate PA1-1 and TNF-a- promoting a pro-coagulant state. Also stimulate tissue factor
- NOS2 – inducible nitric oxide- elevated levels occur in infections and inflammation. Can activate platelets. Inducible nitric oxide, or iNOS, plays a stimulatory role in platelet activation. Platelets generate nitric oxide (NO) in response to agonist stimulation. In particular, there is an iNOS dependent nitric oxide production in platelets induced by thrombin.
- Homocysteine- An amino acid made by methionine in the methylation cycle. It can increase platelet activation, encourage blood clotting and can raise your risk of coronary artery disease, heart attacks, blood clots and strokes.
- Platelet activation – through the activation of endotoxins and cytokines, activated platelets, can activate many coagulation pathways. Activated platelets secrete coagulation factors, expose PS, and support thrombin and fibrin formation.
- Lp(a)- binds up TPA (tissue plasminogen activator)- so high levels can reduce plasminogen and increase blood clotting.
Anti-coagulation definition- slowing down or hindering the process of making clots.
Components of the anti-coagulation process include:
- Plasminogen– a proenzyme that forms plasmin during the process of fibrinolysis.
- Plasmin– a fibrinolytic enzymes that breaks down the cross links between fibrin molecules
- Fibrinolysis – a process that breaks down fibrin blood clots.
- Tissue plasminogen activator (tPA)- enzyme that is involved in dissolution of blood clots. Its primary function includes catalyzing the conversion of plasminogen to plasmin, the primary enzyme involved in dissolving blood clots.
- Anti-thrombin (AT)- a glycoprotein that inhibits the blood clotting pathway- anti-coagulant. Antithrombin is among the number of regulatory mechanisms of the coagulation cascade which provides a counter mechanism to clot formation. It serves as up to 80% of the inhibitory component to thrombin formation, as well as factor IXa and factor Xa inhibition.
- Protein C- a principal inhibitor of coagulation- degrades factor V.
- Protein S- a vitamin K-dependent protease that circulates in plasma at low concentrations and serves a crucial role in the regulation of coagulation. This is an anti-coagulant protein.
- Tissue factor inhibitor (TFPI)- Tissue factor (TF) pathway inhibitor (TFPI) is an anticoagulant protein that inhibits early phases of the procoagulant response.
What is inflammation? This is an important definition as it is a central theme as to the pathology of what could be going on inside causing the upregulation of coagulation.
Inflammation is a cascade event preserved along the evolution from the first multicellular precursor organisms to humans. Its main role is to defend tissues from an insulting agent, such as microbes or direct damage, enabling in most cases a natural return to homeostasis. If inflammation is not someway stopped, it can lead to serious consequences(Valente, Dentoni et al. 2022)
Infections, chemical toxins, mechanical injuries, and many other factors can cause inflammation, a natural defense mechanism (Bagameri, Botezan et al. 2023)
When bacteria invade the organism, host cells are driven to secrete pro-inflammatory cytokines (Bagameri, Botezan et al. 2023)
When the organism is challenged by pathogens, harmful toxins, irradiation, or microbial infections, inflammation occurs as a natural defense mechanism (Bagameri, Botezan et al. 2023).
Numerous physiological and immunological pathways are activated by this complex process; thus, when inflammation becomes dysregulated as a result of certain circumstances, it can harm nearby tissue and induce a wide range of diseases(Bagameri, Botezan et al. 2023)
IL-1β, IL-6, and TNF-α are three major proinflammatory cytokines during the inflammation process (Bagameri, Botezan et al. 2023). COX-2 is the rate-limiting enzyme for converting arachidonic acid to proinflammatory prostaglandins, and it plays a pivotal role in chronic and acute inflammation.
Characteristic inflammatory symptoms, including pain, redness, and heat, involve immune cells, such as B and T lymphocytes, monocytes, macrophages, neutrophils, and basophils, as well as mast and dendritic cells (Bagameri, Botezan et al. 2023).
The inflammatory response is largely influenced by the type of stimulus, all sharing similar mechanisms, such as the identification of pattern-recognition receptors on foreign pathogen’s cell surfaces or intracellular signaling caused by harmed cells or tissues (Bagameri, Botezan et al. 2023)
The first signs of inflammation response are vasodilation and changes in vessel permeability. These factors not only permit the recruitment of cells implied in the inflammatory response but also give substrates for the biosynthesis of important molecules.
Chronic and/or uncontrolled inflammation plays a key role in a variety of diseases (such as cardiovascular diseases, metabolic syndrome, and neurological diseases.
Employing natural remedies as a treatment has received special attention recently because taking anti-inflammatory pharmaceuticals may have a variety of negative side effects.
Key point- inflammation can be an important mediator of the various different processes listed below.
- Fibrinogen is a complex glycoprotein present in high concentrations in plasma.
- Fibrinogen is converted to fibrin, which stabilizes blood clots and promotes hemostasis.
- Fibrin structure and mechanical properties are modified by genetic and environmental factors.
- Fibrin(ogen) also contributes to thrombosis, host defense, inflammation, and wound healing.
What is Fibrinogen?
Fibrinogen is a 340 kDa glycoprotein in human blood plasma that is involved in blood coagulation through its conversion into fibrin by thrombin.
How it happens.
During coagulation, fibrinogen conversion to fibrin occurs via thrombin‐mediated proteolytic cleavage that produces intermediate protofibrils and then mature fibers that provide remarkable biochemical and mechanical stability to clots.
Fibrin formation, structure, and stability are regulated by various genetic, biochemical, and environmental factors. This allows for dynamic kinetics of fibrin formation and structure.
Interactions between fibrinogen and/or fibrin and plasma proteins and receptors on platelets, leukocytes, endothelial cells, and other cells enable complex functions. This process is involved in physiological functions such as hemostasis, thrombosis, pregnancy, inflammation, infection, cancer, and other pathologies.
Disorders in fibrinogen concentration and/or function increase the risk of bleeding, thrombosis, and infection.
Thrombin
Factor IIa, or as commonly known, thrombin, catalyzes the conversion of fibrinogen into fibrin.
Thrombin is a serine endopeptidase naturally produced in humans, and it has a crucial role in the coagulation cascade and the complex process of hemostasis. The liver produces it as an inactivated zymogen, prothrombin, and when the coagulation cascade is activated, this protein is split by other proteases, eventually forming thrombin.
Subsequently, fibrin forms a network of fibrin monomers stabilized by factor XIIIa, this transglutaminase catalyzes the cross-linking of glutamine and lysine residues. The development of a fibrin meshwork provided the appropriate architecture for hemostasis platelet activation, aggregation, and thrombus formation.
What is PAI-1?
Plasminogen activator inhibitor 1 (PAI- 1) eventually terminates the catalytic activity of tPA by binding it. This inactive complex (PAI 1-bound tPA) is removed from the circulation by the liver via the scavenger receptor, LDL receptor-related protein 1 (LRRP1).
In the nervous system, a neuronal-specific inhibitor of tPA, neuroserpin, acts similarly to PAI 1, and the LRRP1 internalizes the inactive tPA-neuroserpin complexes for removal from circulation.
PAI-1 is a very important compound. PAI-1 is an inhibitor of plasminogen. In severe infection, when fibrinolysis is activated, there is an initial activation of plasminogen activation followed by release of PAI-1.
PAI-1 strongly inhibits fibrinolysis causing a net procoagulant situation. The molecular basis is cytokine-mediated activation of vascular endothelial cells; TNFa and IL-1 decreased free tPA and increased PAI-1 production; TNFa increases total uPA production in endothelial cells.
Key point; inflammatory cytokines can increase PAI-1 production- increasing coagulation!
Endotoxins and TNFa stimulate PAI-1 production in liver kidney, lung and adrenals of mice. The net procoagulant state is illustrated by a late rise in fibrin breakdown fragments as seen in experiments with E. coli challenge of baboons.
Factor V
Factor V is a protein of the coagulation system, rarely referred to as proaccelerin or labile factor. In contrast to most other coagulation factors, it is not enzymatically active but functions as a cofactor.
Normally, factor V synthesis principally occurs in the liver. Thrombin activates factor V, and once activated, it will convert prothrombin to thrombin. Activated protein C, one of the principal physiologic inhibitors of coagulation, degrades factor V. In the presence of what is called thrombomodulin, thrombin acts to decrease clotting by activating protein C; therefore, the concentration and the action of protein C are important determinants in the negative feedback loop through which thrombin limits its activation.
Deficiency of Factor V leads to predisposition for hemorrhage, while some mutations (most notably factor V Leiden) predispose for thrombosis.
Endothelial cells
Vascular endothelial cells play a central role in all mechanisms that contribute to inflammation-induced activation of coagulation (Levi, Keller et al. 2003). Endothelial cells respond to the cytokines expressed and released by activated leukocytes. They can also release cytokines themselves. Endothelial cells are also able to express adhesion molecules and growth factors that may not only promote the inflammatory response further but also affect the coagulation response(Levi, Keller et al. 2003). However, it has recently become clear that, in addition to these mostly indirect effects of the endothelium, endothelial cells interfere directly with the initiation and regulation of fibrin formation and removal during severe infection.
Inflammation-induced coagulation activation is characterized by widespread intravascular fibrin deposition, which appears to be a result of enhanced fibrin formation and impaired fibrin degradation.
Enhanced fibrin formation is caused by tissue factor mediated thrombin generation. This occurs simultaneously with a depression of inhibitory mechanisms, such as the protein C and S system.
The impairment of endogenous thrombolysis is mainly due to high circulating levels of PAI-1, the principal inhibitor of plasminogen activation.
The impairments in coagulation and fibrinolysis are mediated by differential effects of various pro-inflammatory cytokines.
Tissue factor
Tissue factor is a protein that is expressed on several cells throughout the body, such as subcutaneous tissue, but not usually direct contact with blood. Through a series of biological activations (such as factor VII and IX and X), prothrombin is activated (Levi, Keller et al. 2003). In cells in contact with blood circulation, TF is induced by the action of compounds such as cytokines, C-reactive protein and advanced glycosylated end products.
The expression of TF on monocytes is stimulated by the presence of platelets and granulocytes- as a result of NFkB activation induced by binding of activated platelets to neutrophils and mononuclear cells (Levi, Keller et al. 2003).
This cellular interaction also markedly enhances the production of IL-1b, IL-8, MCP-1, and TNFa. Under the influence of cytokines, the synthesis of glycosaminoglycans by endothelial cells may be reduced, impairing the inhibitory potential of AT (anti-thrombin).
The good news is the body has several mechanisms that counteract the systemic activation of coagulation that occurs upon inflammation. First, coagulation inhibitors are present to slow down the coagulation mechanism.
Platelets
In addition to the mechanisms described above, platelets may play a role in the pathogenesis of inflammation induced coagulation activation as well. Endotoxin can directly activate platelets.
Many pro-inflammatory cytokines are also capable of inducing platelet activation through the sphingosine pathway and platelet activating factor (PAF) and its receptor. The importance of platelet activation for the activation of coagulation in severe infection is probably related to the provision of a suitable phospholipid surface on the activated platelet membrane for assembly of complexes of activated coagulation factors, such as the prothrombinase complex, consisting of activated factors X and V, prothrombin and calcium. The presence of such a surface may catalyze several-fold the generation of thrombin and render the coagulation system less susceptible to fluid-phase protease inhibitors.
Antithrombin (AT)
Antithrombin is a plasma glycoprotein consisting of 432 amino acid residues integral in the regulation of the coagulation process during bleeding. Antithrombin most notably binds to serine proteases factor II (thrombin), factor IXa, and factor Xa which inhibits the blood clotting process involved in the coagulation cascade pathway. As part of the normal physiological response to bleeding, platelets circulating the plasma become initially activated by multiple factors produced from endothelial cells to aggregate and form a plug. Circulating fibrinogen is then converted into fibrin by thrombin through a series of protease activations, which constitute the reactions of the coagulation cascade pathway. Fibrin acts to stabilize the initial platelet-created plug which determines the completion of the clot formation.
Antithrombin is among the number of regulatory mechanisms of the coagulation cascade which provides a counter mechanism to clot formation. It serves as up to 80% of the inhibitory component to thrombin formation, as well as factor IXa and factor Xa inhibition. Deficiency in antithrombin has clinical links to increased risks of thrombosis, thromboembolism, and associated complications associated with a hypercoagulable state.
What is plasminogen?
Plasminogen is a pro-enzyme (i.e. a zymogen) which is cleaved to form plasmin – also known as fibrinolysin – as part of the fibrinolytic pathway that breaks down fibrin blood clots.
Plasmin, an endogenous fibrinolytic enzyme, breaks the cross-links between fibrin molecules, which are the structural support of the blood clot, and its activity is extremely short-lived. This short duration is because alpha 2-antiplasmin, an abundant inhibitor of plasmin, quickly inactivates it and restricts the action of plasmin to the vicinity of the clot.
Key point- plasminogen is an enzyme that breaks down fibrin- which is what helps break down clots. This pathway is activated when a clot is no longer needed or to prevent a clot from extending beyond the site of injury.
What is tissue plasminogen activator (tPA)?
Tissue plasminogen activator (tPA) is classified as a serine protease (enzymes that cleave peptide bonds in proteins). It is thus one of the essential components of the dissolution of blood clots. Its primary function includes catalyzing the conversion of plasminogen to plasmin, the primary enzyme involved in dissolving blood clots.
The following sequence summarizes the action of tPA:
- tPA attaches to the fibrin on the clot surface.
- It activates the fibrin-bound plasminogen.
- Plasmin is subsequently cleaved from the plasminogen affiliated with the fibrin.
- Plasmin breaks up the molecules of fibrin, and the clot dissolves.
Key point- tPA activates plasminogen. Without it, blood clot dissolution may be impaired.
Tissue factor inhibitor (TFPI)- Tissue factor (TF) pathway inhibitor (TFPI) is an anticoagulant protein that inhibits early phases of the procoagulant response. Alternatively spliced isoforms of TFPI are differentially expressed by endothelial cells and human platelets and plasma. The TFPIβ isoform localizes to the endothelium surface where it is a potent inhibitor of TF–factor VIIa complexes that initiate blood coagulation. The TFPIα isoform is present in platelets.
Protein S- Protein S is a vitamin K-dependent protease that circulates in plasma at low concentrations and serves a crucial role in the regulation of coagulation.
Protein S deficiency is a rare disorder characterized by reduced activity of protein S, a plasma serine protease with complex roles in coagulation, inflammation, and apoptosis. Protein S is an anticoagulant protein. Protein S facilitates the action of activated protein C (APC) on activated factor 5 (F5a) and activated factor 8 (F8a). A deficiency in protein S characteristically demonstrates the inability to control coagulation, resulting in the excessive formation of blood clots (thrombophilia) and venous thromboembolism (VTE)
Protein S deficiency can be congenital or acquired. Mutations in the PROS1 gene cause congenital protein S deficiency
Protein S deficiency is an autosomal dominant pathology. Mutations in a single copy in heterozygous individuals cause mild protein S deficiency, whereas individuals with homozygous mutations present with severe protein S deficiency.
Causes of acquired fluctuations in protein S levels may include:
- Vitamin K-antagonist therapy
- Chronic infections
- Severe hepatic disease
- Systemic lupus erythematosus
- Myeloproliferative disorders
- Nephritic syndrome
- Disseminated intravascular coagulation (DIC)
- The risk of VTE is also increased in patients using oral contraceptives and pregnancy.
Many conditions reduce the blood levels of protein S on both antigenic and functional assays.
These include:
- Vitamin K deficiency
- Liver disease
- Antagonism with warfarin reduces protein S levels.
- Acute thrombosis
- Pregnancy
Almost half of all individuals with protein S deficiency will become symptomatic before age 55.
Please note- your gut bacteria become critical here. Many people do not have enough gut flora to produce vitamin K and this includes Bifidobacteria and many species in Bacteroidetes and Prevotella among others. This is commonly seen when there is dysbiosis, but I can also see this when I see a calcium shell on HTMA and vitamin D deficiency. You need vitamin K to use your Calcium properly, as well as vitamin D.
So why is this a problem?
Infections cause inflammation which triggers fibrin production. Even in the absence of a true infection, inflammation can also trigger fibrin production.
It has been long known that inflammation can lead to the activation of the coagulation system. For example, acute inflammation, as a response to severe infection or trauma, results in a systemic activation of the coagulation system(Levi, Keller et al. 2003). In fact, in the 1990s, it became apparent that cytokines played a mediatory role in the activation of coagulation and subsequent fibrin deposition (Levi, Keller et al. 2003)
The point of impact on the coagulation system was rather the tissue factor-factor VIIa (‘extrinsic’) pathway than the contact system. Tissue factor will be described a bit more below.
One contribution is impairment of the physiological anticoagulant pathway (Levi, Keller et al. 2003). Scientists were able to determine that impaired fibrin removal, by a suppressed fibrinolytic system, contributed importantly to the microvascular deposition of fibrin.
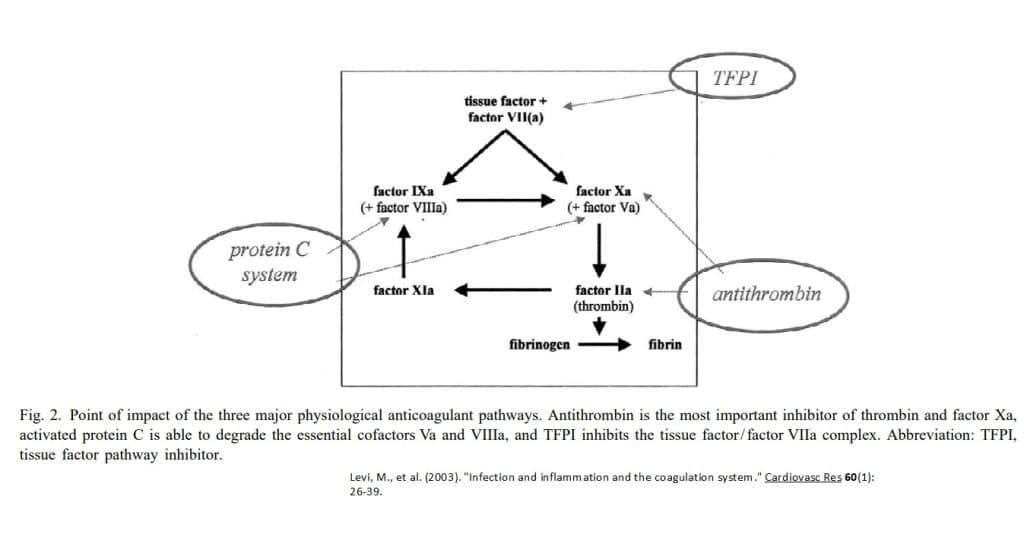
Soluble inhibitors constitute anti- thrombin (AT), proteins C and S, and tissue factor pathway inhibitor (TFPI).
Antithrombin (AT) is thought to be one of the most important inhibitors of the activated coagulation system. Antithrombin inhibits coagulation proteases by 1:1 complex formation, which, in the case of thrombin–antithrombin complexes.
Other components are also involved in fibrinolysis. For example, TNFa is a critical mediator of fibrinolysis as well. The rise of PAI-1 sufficiently blunts fibrinolysis. Antithrombin at tPA released by the endothelium, caused by AT binding to glycosaminoglycans at the endothelial surface.
In experimental models of severe infection, fibrinolysis is activated, demonstrated by an initial activation of plasminogen activation, followed by a marked impairment caused by the release in blood of plasminogen activator inhibitor, type 1 (PAI-1).
PAI-1 strongly inhibits fibrinolysis causing a net procoagulant situation. The molecular basis is cytokine-mediated activation of vascular endothelial cells; TNFa and IL-1 decreased free tPA and increased PAI-1 production, TNFa increases total uPA production in endothelial cells
Endotoxins from bacteria (and yes even your gut bacteria!)
Gut dysbiosis is the disruption in the gut microbiome that is associated with different diseases. Dysbiosis disturbs the gut barrier function, leading to the leakage of harmful metabolites, such as lipopolysaccharides (LPS), and other bacterial components, such as peptidoglycans, into the circulation, which triggers an inflammatory response(El Hage, Al-Arawe et al. 2023)
LPS can stimulate the uptake of modified low-density lipoprotein (LDL) and reduce the efflux of cholesterol from foam cells, promoting monocyte recruitment and macrophage foam cell formation (El Hage, Al-Arawe et al. 2023)
LPS can induce vascular inflammation directly or by producing pro-inflammatory factors from immune cells (El Hage, Al-Arawe et al. 2023)
The increased production of pro-inflammatory cytokines promotes oxidative stress and oxidized LDL (oxLDL), increasing the risk of hypertension via nitric oxide synthase inhibition. This effect reduces vasodilator nitric oxide levels and increases levels of vasoconstrictor endothelin-1 (El Hage, Al-Arawe et al. 2023)
Endotoxins (Lipopolysaccharides, LPS), a major component of the outer membrane of Gram-negative bacteria, are now well as contributors to the inflammation (André, Laugerette et al. 2019)
LPS may originate from skin and mucous membranes or local sites of bacterial infection (André, Laugerette et al. 2019). However, the gut microbiota is considered the main natural reservoir of pro-inflammatory endotoxins in the body (André, Laugerette et al. 2019)
Endotoxins are released when bacteria die, and then dissociated endotoxins are able to cross the gastro-intestinal barrier to end up in the bloodstream(André, Laugerette et al. 2019)
The presence of LPS in the bloodstream is defined as endotoxemia (André, Laugerette et al. 2019)
Briefly, the endotoxic metabolic pathway includes the binding of circulating LPS to LPS-Binding Protein (LBP) and its transfer to the CD14 receptor, which is present both in a membrane-anchored form (mCD14) and in a soluble circulating form (sCD14) (André, Laugerette et al. 2019). The complex LPS–LBP–CD14 initiates the secretion of pro-inflammatory cytokines, such as Interleukin-6 (IL-6) or Tumor Necrosis Factor α (TNFα), through a TLR4-dependent mechanism.
Although LPS is detectable at low concentrations in the circulation of healthy individuals, there is evidence that LPS levels transiently increase following ingestion of fat-rich meals (André, Laugerette et al. 2019). This is defined as as “metabolic endotoxemia”, in contrast to other sources of endotoxemia such as exogenous bacterial infection or sepsis(André, Laugerette et al. 2019) For instance, in mice fed with a four-week high-fat diet, plasma levels of LPS were similar to those observed following a four-week subcutaneous infusion of 300 µg/kg/day of LPS (André, Laugerette et al. 2019).
Metabolic endotoxemia has been proposed as a major cause of inflammation, including chronic low-grade inflammation(André, Laugerette et al. 2019)
Indeed, animal and experimental studies have demonstrated that postprandial state may result in an inflammatory response closely associated with the increase in the circulating levels of LPS (André, Laugerette et al. 2019)
Interestingly, not all dietary interventions lead to increased metabolic endotoxemia and the associated postprandial inflammatory status. Data from animal studies have suggested that dietary fats act as the main macronutrients responsible for postprandial endotoxemia, and that both the quantity and the quality of the dietary fat differentially influence metabolic endotoxemia (André, Laugerette et al. 2019)
According to McFarlin et al (2017), consumption of a single, high-fat, high-calorie meal was associated with an increase in serum endotoxin, triglycerides, metabolic biomarkers, inflammatory cytokines, endothelial microparticles, and monocyte adhesion molecules (McFarlin, Henning et al. 2017)
Moreover, healthy diets rich in unsaturated fatty acids have been associated with lower postprandial circulating levels of LPS, closely associated with lower pro-inflammatory markers. Conversely, the consumption of high-energy or high saturated-fat diets has been associated with increased postprandial levels of LPS and increased circulating levels of pro-inflammatory markers (André, Laugerette et al. 2019)
Currently, the available literature points out that elevated circulating levels of LPS are detrimental for healthy ageing. More specifically, elevated levels of LPS have been associated with a large range of diseases such as obesity, T2DM, coronary artery disease and depression (André, Laugerette et al. 2019)
Endotoxin and TNFa stimulate PAI-1 production in liver kidney, lung and adrenals of mice. (Levi, Keller et al. 2003) The net procoagulant state is illustrated by a late rise in fibrin breakdown fragments as seen in experiments with E. coli challenge of baboons.
Endotoxin is the lipopolysaccharide compound from Gram-negative bacteria inducing the sepsis syndrome and disseminated intravascular coagulation (DIC) (Levi, Keller et al. 2003) . DIC is a rare by serious condition that causes abnormal blood clotting throughout the body’s blood vessels. You may develop DIC if you have an infection or injury that affects the body’s normal blood clotting process.
Gram-negative bacteria liberate endotoxins from their membrane which interact with cell surfaces by different pathways. In blood, endotoxin directly binds to CD 14 at monocytes, and binds to endothelial cells after complexing with lipopolysaccharide binding protein (LBP) and the Toll-like receptor 4 complex(Levi, Keller et al. 2003). By these interactions endotoxin induces a number of signaling pathways leading to the activation of the NFkB system, which starts the transcription of genes of proinflammatory cytokines, TF and others. Except for a direct positive effect of endotoxin on TF synthesis, the formation of the cytokines IL-1, TNF-a, and IL-6 stimulates TF formation ( Levi, Keller et al. 2003.
Key point- endotoxins from gram-negative bacteria can stimulate the formation of cytokines that then stimulate tissue factor- starting up the coagulation pathway.
This has been observed, for example, after intravenous endotoxin challenge. A rapid production and liberation of proinflammatory cytokines was observed. Among those, TNFa and IL-6 are important for inducing fibrinolytic and procoagulant changes in the blood. IL-6 is an important mediator of procoagulant effects, while TNFa is particularly involved in the fibrinolytic response to endotoxin.
Studies in baboons challenged with lethal amounts of E. coli (a model of Gram-negative septicemia) have underscored the importance of cytokines in mediating the sepsis syndrome, and demonstrated the prolonged proinflammatory course in which the actual formation of fibrin is particularly evident at 24 h after E. coli challenge(Levi, Keller et al. 2003) .
Both in the chimpanzee endotoxin model and the E. coli model, TF is essential for inducing coagulation activity, and inhibition of the TF pathway abolishes clotting activation.
As mentioned earlier, fibrinolytic activity is markedly regulated by PAI-1, the principal inhibitor of this system. Recent studies have shown that a functional mutation in the PAI-1 gene, the 4G/5G polymorphism can influence the plasma levels of PAI-I. Patients with the 4G/4G genotype had significantly higher PAI-1 concentrations in plasma and an increased risk of death.
These studies are the first evidence that genetically determined differences in the level of fibrinolysis influence the risk of developing complications of a Gram-negative infection. Consequently, PAI-1 is also a positive acute phase protein. Therefore, a higher plasma concentration may also be a marker of disease rather than a causal factor. Interestingly platelet granules contain large quantities of PAI-1 and release PAI-1 upon their activation as well. Since platelets become activated in case of severe inflammation and infection, this may further increase the levels of PAI-1 and contribute to the fibrinolytic shut-off.
This problem is enhanced in advanced age.
Aging leads to several changes in cells, tissues, and organs and is influenced by an individual’s genetics, lifestyle, and environment (El Hage, Al-Arawe et al. 2023)
. The term “immunoscence” first appeared a few decades ago to refer to impaired or faulty immune responses leading to a decrease in the ability to trigger the immune response and effectively produce antibodies against different pathogens (El Hage, Al-Arawe et al. 2023)
The gut microbiome undergoes dynamic changes through time, and gut dysbiosis is an age-related complication caused by host senescence, changes in nutritional behavior, drug use, and the lifestyle of aged people (El Hage, Al-Arawe et al. 2023)
The changes in the gut microbiome include shifts in bacterial composition and metabolic function (El Hage, Al-Arawe et al. 2023)
In humans, age-related gut dysbiosis is characterized by increased inter-individual variation and decreased species diversity; specifically, a loss of Clostridiales and Bifidobacterium, an enrichment of Proteobacteria, Lactobacilli, and an overrepresentation of pathobionts such as Enterobacteriaceae (El Hage, Al-Arawe et al. 2023)
However, the major gut microbiota aging feature is the decreased ratio of Firmicutes/Bacteroidetes (El Hage, Al-Arawe et al. 2023)
Schneeberger et al. reported that aged mice showed a decrease in beneficial gut bacteria, such as in Clostridium members of cluster IV that produce SCFAs and Akkermansia muciniphila, and an increase in pro-inflammatory microbes (El Hage, Al-Arawe et al. 2023)
Overall, the decrease in intestinal commensal microbes diversity is associated with increased susceptibility to pathogen infection accompanied by disturbance of the gut mucosal barrier and enrichment in pro-inflammatory cytokines(El Hage, Al-Arawe et al. 2023)
All these events have a detrimental consequence in aging. Recent studies suggested that gut dysbiosis is associated with the development of several chronic diseases including cardiovascular disease and other metabolic disorders (El Hage, Al-Arawe et al. 2023)
Many studies have reported a close relationship between TMAO levels, aging, and age-related diseases. Several animal models have been used to identify mechanisms that underlie TMAO’s role in senescence. Cell senescence involves many processes including increased production of reactive oxygen species (ROS), mitochondrial dysfunction, and senescence-associated secretory phenotype (SASP) (El Hage, Al-Arawe et al. 2023)
Many potential mechanisms underlie TMAO’s role in aging, including the inhibition of sirtuin 1 (SIRT1) expression, which increases oxidative stress and results in the activation of the p53/p21/Rb pathway. Increased P53 and P21 acetylation and reduced CDK2, cyclinE1, and Rb phosphorylation are followed by enhanced endothelial cell senescence and vascular aging. In addition, TMAO increases the accumulation of ROS, matrix metalloproteinase 2 (MMP2), and matrix metalloproteinase 9 (MMP9) in vivo and in vitro, which are associated to oxidative stress in cells. Furthermore, high TMAO levels are linked to increased expression of pro-inflammatory cytokines, such as TNF-α and IL-1β, as well as decreased production of anti-inflammatory cytokines such as IL-10 (El Hage, Al-Arawe et al. 2023).
Mast Cell Activation
Mast cells (MCs) are hematopoietic tissue immune cells that act both as effector and regulatory cells in adaptive and innate immunity (Seidel, Hertfelder et al. 2021).
Traditionally, MCs have been referred to as the key effector cells of allergic diseases, such as urticaria, allergic rhinitis and bronchial asthma (Seidel, Hertfelder et al. 2021). MCs are mostly located in the border between host and environment such as skin, gastrointestinal and respiratory mucosa and thus may have easy contact with the external environmental pathogens (Yu, Song et al. 2021).
This versatility is reflected in the myriad of immunologic and non-immune activation stimuli resulting in the secretion of a large number of pre-stored mediators, such as histamine and tryptase, and numerous de novo-synthesized lipid mediators (e.g., eicosanoids), chemokines, and cytokines (Seidel, Hertfelder et al. 2021).
MC activation may affect hemostasis by vascular and cellular components, such as platelets, monocytes and neutrophils, as well as clotting and fibrinolytic factors (Seidel, Hertfelder et al. 2021)
Primary MC disease comprises a group of historically defined different disease entities (consisting of several variants (Seidel, Hertfelder et al. 2021)
- systemic mastocytosis (SM)
- MC activation syndrome (MCAS)
- cutaneous mastocytosis (CM)
- MC sarcoma and hereditary alphatryptasemia (HAT).
The prevalence of MCAS, at least in Germany, is about 17% and about 20% in the U.S. , and that of HAT was found to be 4–6% of the general population hence both being common disorders (Seidel, Hertfelder et al. 2021). SM, CM, and MC sarcoma are rare.
A new view of the development and categorization of primary MC disease in that it is one polygenic multifactorial disease entity characterized by epigenetic and genetic alterations (somatic and germline mutations) in a variety of genes disorders (Seidel, Hertfelder et al. 2021). The combinatorial calculated number of possible combinations of the genetic alterations suggests that each patient affected by a primary MC disease has a unique mutational pattern or profile driving a unique pattern of aberrant MC mediator production and release disorders (Seidel, Hertfelder et al. 2021).
Due to both the widespread distribution of MCs in the organism and the great heterogeneity of aberrant mediator expression patterns, symptoms can involve virtually all organs and tissues; thus, the clinical presentation of primary mast cell disease is very diverse disorders (Seidel, Hertfelder et al. 2021).
Severity of symptoms, too, in one tissue/organ/system can vary substantially from one patient to the next, at different times in an individual patient, or even in different systems at the same time in an individual patient disorders (Seidel, Hertfelder et al. 2021).
Mast cells, Hemostasis and Thrombosis
MCs are mostly located in the border between host and environment and thus may have easy contact with the external environmental pathogens (Yu, Song et al. 2021). These cells express receptors which can recognize pathogen-associated molecular patterns such as Toll-like receptors (TLR2/4) and C-type Lectins receptors (Dectin-1/2). (Yu, Song et al. 2021). Currently, more and more data indicate that MCs can be interacted with some fungi (Candida albicans, Aspergillus fumigatus and Sporothrix schenckii). (Yu, Song et al. 2021)
It is demonstrated that MCs can enhance immunity through triggered degranulation, secretion of cytokines and chemokines, neutrophil recruitment, or provision of extracellular DNA traps in response to the stimulation by fungi (Yu, Song et al. 2021)
Interestingly, findings indicate that MC-released histamine may either cause or inhibit thrombosis, depending on whether it acts on resting endothelial cells or on cells pre-activated by other inflammatory stimuli (Seidel, Hertfelder et al. 2021)
The array of substances released by activated MCs which may influence hemostasis include:
- histamine
- Heparin
- vasoactive intestinal polypeptide (VIP)
- prostanoids
- the proteases tryptase and chymase
- tissue-type plasminogen activator (tPA
- factor VIII
Since high concentrations in circulation and other tissues of these mediators can be achieved by their release from MCs, both clinically significant bleeding and thrombosis can occur in MC disease (Seidel, Hertfelder et al. 2021)
Platelet activation may trigger MC activation, and hence stimulate MC mediator release, contributing to distortion of hemostasis (Seidel, Hertfelder et al. 2021)
Mast cell mediators have both pro and anti-coagulant activity(Seidel, Hertfelder et al. 2021). For example, 50% of patients with MC disease exhibit clinical signs of bleeding diatheses(Seidel, Hertfelder et al. 2021).
The presence of hemorrhagic disorders in patients with MC disease is mainly explained by the anticoagulant activity of MC degranulation products like heparin, histamine and tryptase and by hyperfibrinolysis (Seidel, Hertfelder et al. 2021).
In human tissues, mast cells are the main source of heparin(Seidel, Hertfelder et al. 2021). The anticoagulant effect of heparin consists of binding to antithrombin leading to inactivation of thrombin and factor Xa (Seidel, Hertfelder et al. 2021).
Endothelial cells and MCs represent an important source of tissue type plasminogen activator (tPA), indicating that MC activation plays an important role in endogenous fibrinolysis (Seidel, Hertfelder et al. 2021)
The release of tPA from endothelial cells might also lead to an early destabilization of hemostatic clots by fibrinolysis(Seidel, Hertfelder et al. 2021).
Studies to date suggest that the main causes of bleeding diatheses in primary MC disease likely include pathological hyperfibrinolysis in MCAD with elevation of tPA levels released in significant amounts also from MCs and endothelial cells as well without concomitant release of plasminogen activator inhibitor type 1 (PAI) (Seidel, Hertfelder et al. 2021).
The important MC mediator histamine can influence hemostasis by upregulating thrombomodulin activity in endothelial cells and thereby promoting the activation of the protein C/S (PC/S) system by thrombomodulin-bound thrombin (Seidel, Hertfelder et al. 2021)
The PC/S system is a major anticoagulant that is required for the downregulation of blood coagulation(Seidel, Hertfelder et al. 2021). Moreover, histamine also stimulates endothelial cells to release VWF and tPA (Seidel, Hertfelder et al. 2021)
Another MC mediator, prostaglandin D2, is known to inhibit platelet aggregation via activation of platelet adenylate cyclase (Seidel, Hertfelder et al. 2021)
MC tryptase could have opposite effects on hemostasis as both an anticoagulant and a procoagulant (Seidel, Hertfelder et al. 2021). The antithrombotic function is mainly due to the ability of tryptase to proteolytically degrade fibrinogen, predominantly by cleavage of the C-terminal α-chains before thrombin can convert fibrinogen to fibrin and subsequently impair fibrin polymerization (Seidel, Hertfelder et al. 2021). Tryptase also acts directly on the fibrinolytic pathway by activating the uPA resulting in the direct, i.e., fibrin-independent conversion of plasminogen into plasmin. Therefore, uPA promotes the proteolytic degradation of fibrinogen and other clotting factors(Seidel, Hertfelder et al. 2021).
In this context, it can be speculated whether the intracerebral bleeding which occurs in about 0.6% of patients with Long Covid syndrome might be linked to dysfunctional MCs in patients with MC disease (Seidel, Hertfelder et al. 2021). Meanwhile it has been revealed that COVID-19 disease is predominantly a vascular disease, leading to leaky blood vessels (Seidel, Hertfelder et al. 2021)
In parallel with inducing a bleeding diathesis, MCs are thought to contribute to venous thromboembolism and atherosclerosis through release of granular constituents including :
- histamine
- Prostanoids
- heparin (by its activation of factor XII (FXII))
- cytokines, the proteases tryptase and chymase (via activation of protease-activated receptors and clotting factors such as fibrinogen, FXII, and XIII)
- platelet activating factor (PAF, which activates platelets and leads to fibrin formation via Factor XII activation)
- secretion of VWF
- Factor VIII (which is also present in MCs [61,62])
- soluble P-selectin
- and increased intercellular adhesion molecule-1 (ICAM-1) expression.
FXII activation by heparin and/or anionic polyphosphates initiates the intrinsic pathway of coagulation(Seidel, Hertfelder et al. 2021).
MCs also release extracellular traps, and the presence of MC-derived traps has been reported in coronary thrombi which stimulate thrombosis (Seidel, Hertfelder et al. 2021).
MCs also generate growth factors, leukotrienes, prostaglandins, and nitric oxide NO. They can be activated by IL1, IL-33, IL-9, tryptase, and certain neuropeptides; on the other hand, cytokines such as IL-33 and IL-1 may activate MC without degranulation (Kritas, Gallenga et al. 2018).
Mast cells are present in tissues, especially the skin, where they participate in both the initial and late immune phases producing cytokines and granular proteins(Kritas, Gallenga et al. 2018).
Fungi-Mast cell Connection
Mast cells (MCs) have been considered as the core effector cells of allergic diseases. However, there are evidence suggesting that MCs are involved in the mechanisms of fungal infection (Yu, Song et al. 2021)
Molds include all species of microscopic fungi, the spores of which are small molecules, ubiquitous, mostly found in soil with higher rainfall and high humidity, in the atmosphere of urban and rural settings and in decaying vegetation (Kritas, Gallenga et al. 2018)
Fungal infections are a serious health problem all over the world. Currently, more than 300 million people suffer from severe fungal infections, and an estimated over 1.5 million people die from deep fungal infections each year Yu, Song et al. 2021). With broad-spectrum antibiotics used and the increase of immunodeficiency disease such as acquired immune deficiency syndrome, pathogenic fungi opportunistic infections showed a trend of rising sharply Yu, Song et al. 2021)
Mold exposure to immune compromised subjects may lead to an increased risk of lung infection, and increase the risk of childhood asthma(Kritas, Gallenga et al. 2018). Inhalation is considered the primary way that people are exposed to fungi, along with skin and diet. However, molds are generally not harmful to healthy humans(Kritas, Gallenga et al. 2018).
According to the Centers for Disease Control and Prevention, some of the most common indoor molds are Cladosporium, Penicillium, Aspergillus, Alternaria, Stachybotrys chartarum (Seidel, Hertfelder et al. 2021)
Candida
C. albicans is one of the most common dimorphic fungus colonizing mucosal surfaces such as gastrointestinal tract,oral-nasal cavity and skin(Yu, Song et al. 2021)
Vulvovaginal candidiasis is one of the most common forms of candidiasis(Yu, Song et al. 2021)
When host immunity is suppressed or damaged, C. albicans can result in severe invasive diseases(Yu, Song et al. 2021). Mast cells activated by Candida Albicans release IL-8 which is a chemokine that attracts neutrophils.
Invasive candidiasis is the most common critical care-associated fungal infection in patients hospitalized in intensive care units, with mortality rates between 40% and 55% (Yu, Song et al. 2021)
MCs play a positive role in the defense mechanism against C. albicans infections, including phagocytosis and killing yeasts in the extracellular environment (Yu, Song et al. 2021)
Both yeast and hyphae of C. albicans can induce the degranulation of bone marrow derived mast cells (BMMCs) and lead to the production of different cytokines and chemokines (CCL3 and CCL4) that regulate immune response (Yu, Song et al. 2021)
In response to the stimulation by yeast cells, BMMCs showed increased production of IL-6 and IL-b that could not be induced by hyphae (Yu, Song et al. 2021). Reactive oxygen species and nitric oxide are released as defense by mast cells.
Some studies have found that mucosal mast cells (MMCs) contribute to barrier function loss in leaky gut models or higher sensitization against food antigen in response to yeast stimulation(Yu, Song et al. 2021). Whereas CTMCs induce local protective tolerance to infection by release of anti-inflammatory cytokine in response to hyphae (Yu, Song et al. 2021)MCs promoting either inflammatory dysbiosis or tolerance were also observed in vulvovaginal candidiasis (Yu, Song et al. 2021).
Aspergillus
Aspergillus is the most common fungus group in our environment and can cause several pathological disorders; however, in individuals with normal functioning of the immune system, infections caused by Aspergillus(Kritas, Gallenga et al. 2018). After Candida, which causes opportunistic mycosis, Aspergillus species can cause different clinical situations such as opportunistic infections, allergies and toxicity. It is the most commonly isolated invasive infection (Kritas, Gallenga et al. 2018)
Epithelium toll-like receptors react with Fungi, generating inflammatory cytokine transcription and translation. The pro-inflammatory TLR signaling pathway leads to generation of inflammatory cytokines and chemokines, including TNF and interferon regulatory factors via NFK-B activation (Kritas, Gallenga et al. 2018)
Fungi such as Aspergillus and Candidiasis can provoke allergy by increasing the levels of IgE and number of mast cells involved with an increase of cytokines such as IL-4, Il6-, IL-1, IL-2, INF-gamma(Kritas, Gallenga et al. 2018)
. The recruitment of mast cells into fungus-infected tissue is a crucial feature of allergic inflammation where cytokines and chemokines are generated. Therefore, fungus are involved in allergic fungal asthma which is a chronic disorder mediated by mast cells(Kritas, Gallenga et al. 2018)
In general, mold has an interaction with the human immune system leading to activation of innate immune cells following the release of pro-inflammatory cytokines such as IL-1(Kritas, Gallenga et al. 2018). IL-1 family members react to pathogens and promote an innate immune response- but many are highly inflammatory(Kritas, Gallenga et al. 2018). IL-1 is found in individuals with mold inflammation as it is thought to contribute to leukocyte migration, tissue remodeling, pain, arachidonic acid cascade and activation and inflammation(Kritas, Gallenga et al. 2018) It has been reported that IL-1 activates mast cells and promotes generation of chemokines and therefore cell recruitment influencing allergic mediator release
Biofilms
Uropathogenic Ecoli and Biofilm formation
E Coli UTI’s are very common among patients with IC. Uropathogenic Escherichia coli (UPEC) is the most common cause of UTI, accounting for approximately 80% of infections(Eberly, Floyd et al. 2017)
UPEC readily forms multi-cellular communities known as biofilms on the surface of catheter materials, bladder walls, as well as within bladder epithelial cells.
The formation of biofilms markedly impedes the treatment of UTIs by protecting encased bacteria from both the host immune response and antimicrobial therapy. Bacteria are in close proximity within the biofilm, facilitating the exchange of genetic material, such as antimicrobial resistance plasmids and transposons(Eberly, Floyd et al. 2017)
To form a biofilm, bacteria must first attach to a surface. Adherence of UPEC strains can be influenced by a wide variety of intrinsic factors, such as adhesive proteins, fibers, and exopolysaccharide molecules; the carriage and expression of such factors differs from strain to strain (Eberly, Floyd et al. 2017)
Once attached to the surface bacteria change from a planktonic form to a sessile form, which replicate while producing extracellular matrix (ECM). This ECM encases the bacteria in a micro-colony. The ECM can be made up of one or more components including exopolysaccharides, proteinaceous material, and extracellular DNA (Eberly, Floyd et al. 2017)
As the biofilm colony grows and matures, bacteria within the ECM respond to signals from their surrounding environment, eventually leading to a portion of the encased bacteria dispersing from the biofilm colony (Eberly, Floyd et al. 2017)
The dispersed bacteria can return to the planktonic form or continue the process of biofilm formation elsewhere. Alternatively, the bacteria can form quiescent reservoirs in the urothelium which are thought to contribute to recurrent infection(Eberly, Floyd et al. 2017)
One of the most critical adherence factors identified in UPEC is a class of fibers termed type 1 pili (Eberly, Floyd et al. 2017)
Type 1 pili mediate UPEC adhesion to bladder epithelial cells, and their deletion greatly diminishes biofilm formation under laboratory and in vivo. Many things can affect this process, such as availability of oxygen, urine pH, constituents in the urine such as TMAO (more information below). But one important point is that increased fibrin can also increase biofilms.
Key point- Increased fibrin, no matter the source, is associated with biofilm formation!
Pair that with coagulation defects and it is a recipe for a perfect storm!
The ability of bacteria to form biofilms on host surfaces is a crucial virulence factor and protects them against innate host defenses and antimicrobial agents. The formation of biofilms is important in the pathogenesis of several subacute and chronic human bacterial infections.
There was an interesting study in 2013, in which scientists reported that fibrinogen induced biofilm formation and adherence to endothelial cells with Streptococcus species (Bedran, Azelmat et al. 2013).
In the study, they showed that S. mutans biofilm formation on a polystyrene surface is promoted by plasma and that fibrinogen is the component responsible for this effect.
Jung et al. previously reported that platelets are required for S. mutans biofilm formation in plasma, while this study showed that fibrinogen alone is sufficient. In this study, S. mutans growing in a fibrinogen-induced biofilm was found to be more resistant to the bactericidal activity of penicillin. This enhanced resistance to penicillin was likely related to the stable architecture of the biofilm, which restricted the penetration of the antibiotic. The presence of persister cells (dormant bacteria) may also explain the resistance of S. mutans in biofilms to killing by penicillin, which acts on growing cells(Bedran, Azelmat et al. 2013).
Fibrinogen also increased the adherence of S. mutans to endothelial cells. Since S. mutans is known to invade human coronary artery endothelial cells and that close interactions between bacteria and host cells are critical in the invasive process, this fibrinogen-induced adhesion may enhance the ability of S. mutans to enter endothelial cells and avoid being eliminated by the immune system(Bedran, Azelmat et al. 2013).
To identify the mechanism by which fibrinogen promotes biofilm formation by S. mutans, they evaluated the ability of this species to convert fibrinogen into fibrin and to bind fibrinogen to its cell surface. While S. mutans could not mediate the conversion of fibrinogen into fibrin, it possessed a fibrinogen-binding activity. This property may allow bacteria to attach to each other through fibrinogen-mediated cross bridging. Fibrinogen binding to streptococci also plays a significant role in enabling them to adhere to host surfaces and in protecting them from the host immune system(Bedran, Azelmat et al. 2013).
The diet-dysbiosis- biofilm link: TMAO
As mentioned earlier, various constituents in the urine can affect the microbiome of the bladder. A journal article published in 2022 (Qiao, et al) reported that TMAO can reduce the susceptibility of E coli to multiple antibiotics.
Trimethylamine N-oxide (TMAO), an important intestinal flora-derived metabolite, plays a role in the development of cardiovascular disease and tumor immunity (Qiao, Liang et al. 2022)
Trimethylamine N-oxide (TMAO) is a small colorless amine oxide generated from choline, betaine, and carnitine by gut microbial metabolism (Velasquez, Ramezani et al. 2016)
TMAO is formed from trimethylamine (TMA), which is generated by the action of gut microbiota from dietary choline and phosphatidylcholine (lecithin) (Velasquez, Ramezani et al. 2016)
Red meat, eggs, dairy products and salt-water fish are rich in choline, lecithin, and carnitine and, hence, are a potential source of TMAO(Velasquez, Ramezani et al. 2016).
In humans, a mutation of the FMO3 causes trimethylaminuria, a condition wherein individuals excrete TMA, rather than TMAO. This condition known as fish odor syndrome is characterized by a fishy body odor in urine, sweat, breath and other bodily excretions (Velasquez, Ramezani et al. 2016)
It accumulates in the tissue of marine animals in high concentrations and protects against the protein-destabilizing effects of urea (Velasquez, Ramezani et al. 2016). Plasma level of TMAO is determined by a number of factors including diet, gut microbial flora and liver flavin monooxygenase activity (Velasquez, Ramezani et al. 2016).
Human and animal studies suggest that several families of bacteria are involved in TMA/TMAO production, namely, Deferribacteraceae, Anaeroplasmataceae, Prevotellaceae and Enterobacteriaceae (Velasquez, Ramezani et al. 2016). Recent studies using isolates of commensal bacteria in the human intestine have identified nine strains capable of producing TMA from choline in vitro: eight species representing two different phyla (Firmicutes and Proteobacteria) and six genera that showed significant choline consumption and TMA production including Anaerococcus hydrogenalis, Clostridium asparagiforme, C. hathewayi, C. sporogenes, Escherichia fergusonii, Proteus penneri, Providencia rettgeri, and Edwardsiella tarda (Velasquez, Ramezani et al. 2016).
Recently, experiments with the choline-degrading sulfate reducing bacterium Desulfovibrio desulfuricans identified a gene cluster (including cutD) involved in radical C–N bond cleavage of choline leading to generation of TMA.
Previous studies also described that many human gut colonizing bacteria are capable of producing TMA, which leads to an increase of TMAO levels in plasma. These gut bacteria include Streptococcus sanguinis, Desulfovibrio alaskensis, Desulfovibrio desulfuricans, Acinetobacter, Serratia, Escherichia coli, Citrobacter, Klebsiella pneumoniae, Providencia, Shigella, Achiomobacter, and Sporosorcine, which belong to the Firmicutes and Actinobacteria phyla(El Hage, Al-Arawe et al. 2023). On the other hand, bacteria belonging to phylum Bacteroidetes are not able to produce TMA (El Hage, Al-Arawe et al. 2023)
Several previous studies have investigated the impact of host factors, such as diet and dietary compounds, on TMAO plasma levels, and it was reported that higher plasma TMAO levels have been linked to an animal-based diet compared to vegetarians. (El Hage, Al-Arawe et al. 2023)
Advancing age has also been strongly linked to TMAO levels. As age increases, the host’s physiology and function are altered. For example, the epithelial integrity of the colon, which is needed to promote the influx of bacterial metabolites, including TMAO, might be reduced. Rath et al. reported that there was an association between carotid intima-media thickness (IMT) and TMAO only in individuals above 65 years of age, which indicates that aging people are principally affected by this metabolite (El Hage, Al-Arawe et al. 2023)
In humans, a positive correlation between elevated plasma levels of TMAO and an increased risk for major adverse cardiovascular events and death is reported (Velasquez, Ramezani et al. 2016). The atherogenic effect of TMAO is attributed to alterations in cholesterol and bile acid metabolism, activation of inflammatory pathways and promotion foam cell formation (Velasquez, Ramezani et al. 2016). TMAO levels increase with decreasing levels of kidney function and is associated with mortality in patients with chronic kidney disease (Velasquez, Ramezani et al. 2016).
Trimethylamine N-oxide (TMAO) is a bioactive molecule produced by gut microbial-derived metabolism (Qiao, Liang et al. 2022).
In humans, diets rich in TMA precursors (e.g., choline and L-carnitine) are the main source of TMAO.
The intestinal flora converts dietary nutrients to TMA, which is absorbed into the bloodstream through the intestinal mucosa and then converted to TMAO in the liver by flavin-containing monooxygenases (FMOs).
There are some associations with TMAO levels and increased risk of diseases such as (Qiao, Liang et al. 2022)
- Diabetes
- Heart failure
- Atherosclerosis
- IBD
- Alzheimer’s disease
TMAO and antibiotic resistance
Recently, an interesting study has found that a high-fat diet leads to dysbiosis of intestinal flora and depletion of the microbial metabolite indole-3-acetic acid (IAA), leading to reduced antibiotic efficacy against bacterial infections(Qiao, Liang et al. 2022).
The study by Oliver et al. found that a diet high in fiber and low in animal protein established an association with antibiotic resistance by shaping the human gut microbiota(Qiao, Liang et al. 2022).
The intestinal microbiota carries a large number of antibiotic resistance genes, and those metabolites that reduce antibiotic susceptibility may exacerbate the emergence and spread of resistant bacteria(Qiao, Liang et al. 2022).
The inhibition of antibiotic efficacy by intestinal microbiota metabolites may drive the enrichment and evolution of antibiotic-resistant bacteria. If so, bacterial resistance will become even more problematic. Because the influence of diet on intestinal metabolites is extensive (, the dietary structure of different individuals is complex. This makes it particularly important to explore the effect of intestinal flora metabolites on antibiotic efficacy.
Interestingly, TMAO reduced the susceptibility of Escherichia coli to a variety of antibiotics(Qiao, Liang et al. 2022). Further data suggest that TMAO does not rely on activation of the TMAO sense-regulatory system and the classical multidrug efflux system to mediate the inhibition of antibiotic efficacy. TMAO enhanced the survival of E. coli under lethal urea (protein denaturant) stress and also protected E. coli from killing by two disinfectants (ethanol and H2O2) that cause protein damage(Qiao, Liang et al. 2022). In conclusion, their study suggests that TMAO has the potential to improve bacterial survival under anti-biotics and other lethal stresses through its protective effect against protein denaturation/damage(Qiao, Liang et al. 2022).
The plasma TMAO levels show wide inter- and intra-individual variations. Circulating levels of TMAO are determined by a number of factors, including diet, gut microbial flora, liver FMO enzymes and kidney function (Velasquez, Ramezani et al. 2016).
Elevated TMAO levels are strongly associated with degree of renal function (Velasquez, Ramezani et al. 2016).
TMAO generated by gut microbiome exacerbates impaired glucose tolerance, inhibits hepatic insulin signaling, and promotes adipose tissue inflammation in mice that are maintained on a high-fat high-sugar diet (Velasquez, Ramezani et al. 2016).
The mechanism by which TMAO promotes atherosclerosis also remains speculative. The proposed mechanisms include changes in cholesterol and sterol metabolism, promotion of foam cell formation by increasing expression of scavenger receptors on macrophages, and causing alternations in bile acid metabolism and sterol transporters in the liver and intestine (Velasquez, Ramezani et al. 2016).
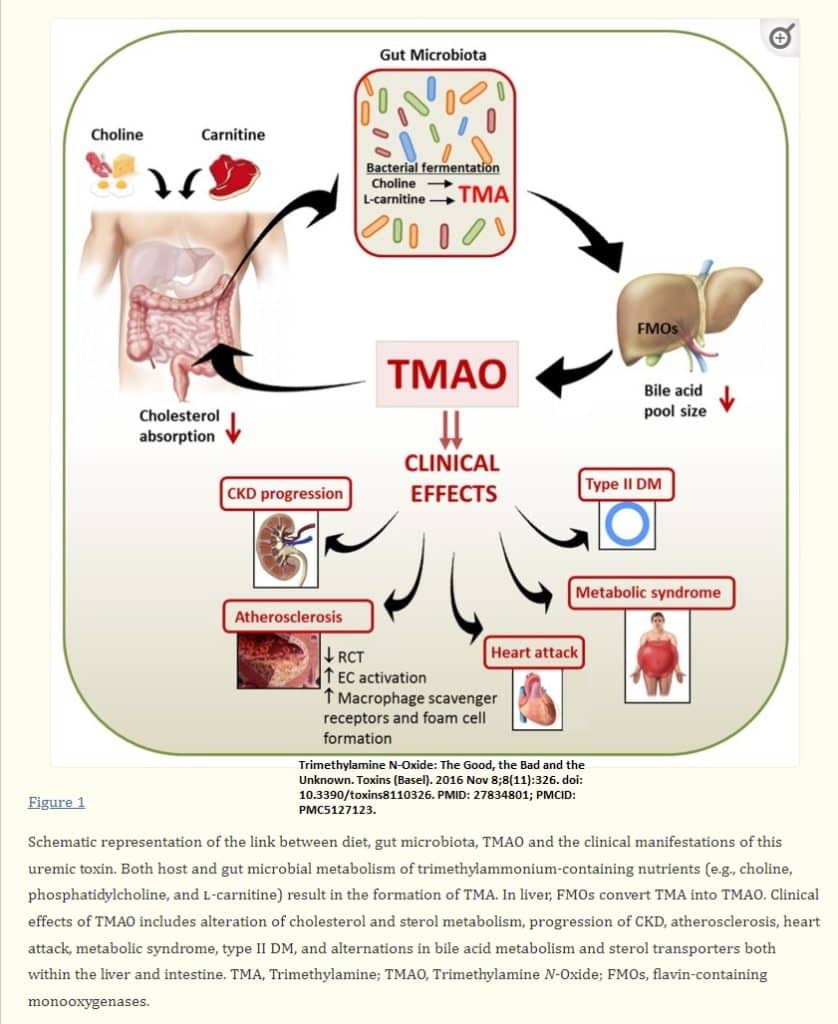
TMAO and thrombosis risk
Both in vivo studies in mice and in vitro studies in cultured human aortic endothelial and vascular smooth muscle cells have shown that physiological levels of TMAO induce expression of inflammatory cytokines and adhesion molecules(Velasquez, Ramezani et al. 2016).
This activation was mediated, in part, by the NF-κB signaling pathway (Velasquez, Ramezani et al. 2016).
In a series of studies in animals employing dietary choline, or TMAO, scientists showed that gut microbe-derived TMAO enhances platelet responsiveness and thrombosis in vivo(Velasquez, Ramezani et al. 2016).
In addition, studies in humans also showed that plasma TMAO levels independently predicted incident thrombosis (heart attack, stroke) risk(Velasquez, Ramezani et al. 2016).
Direct exposure of platelets to TMAO enhanced sub-maximal stimulus-dependent platelet activation from multiple agonists through augmented Ca2+ release from intracellular stores(Velasquez, Ramezani et al. 2016).
Collectively, these findings provide evidence for a mechanistic link between gut microbes, platelet function, and thrombosis risk (Velasquez, Ramezani et al. 2016).
Wang et al. showed that diet containing choline, TMAO or betaine could enhance up-regulation of multiple macrophage scavenger receptors that are linked to atherosclerosis (Velasquez, Ramezani et al. 2016).
In mice with an intact intestinal microbiota, dietary supplementation with TMAO, carnitine or choline reduced in vivo reverse cholesterol transport (Velasquez, Ramezani et al. 2016). Also, mice supplemented with dietary TMAO showed a significant reduction in cholesterol absorption, as well as liver expression of bile acid synthetic enzymes cytochrome P450, family 7, subfamily a, polypeptide 1 (Cyp7a1) and cytochrome P450, family 27, subfamily a, polypeptide 1 (Cyp27a1) (Velasquez, Ramezani et al. 2016). The bile acid pathway plays a major role in cholesterol elimination and blocking this pathway may promote atherogenesis(Velasquez, Ramezani et al. 2016).
Endometriosis
A lot of my patients have endometriosis. Interesting a study published in 2018 by Ding et al reported that platelets play a crucial role in the development of endometriosis (Ding, Liu et al. 2018). In addition, women with endometriosis appear to be in a state of hypercoagulability(Ding, Liu et al. 2018)
Endometriosis, defined as the deposition and growth of endometrium-like tissues outside the uterine cavity, is a common disorder affecting about 6% to 10% of reproductive-age women(Ding, Liu et al. 2018)
As a major contributor to pelvic pain and infertility, it is a leading cause of gynecological hospitalization in the United States and likely in many other parts of the world.
Endometriosis has been traditionally viewed as a hormonal disease. This is often attributed to both estrogen-dependent growth and maintenance of ectopic endometrium and increased local production of estrogens owing to aberrant steroidogenesis. Equally important, it is also conceptualized as a pelvic inflammatory condition, featuring increased production of proinflammatory cytokines and chemokines(Ding, Liu et al. 2018)
In the past 2 decades, however, it is becoming increasingly evident that inflammation and coagulation—long regarded as 2 separate entities—are 2 major host–defense systems that interact with each other (Ding, Liu et al. 2018)
In fact, the 2 entities are intricately entwined: Inflammation activates the coagulation cascade, and coagulation modulates the inflammatory activity in many ways as in cardiovascular disease.
Activated platelets are found to play a critical role in initiating inflammation(Ding, Liu et al. 2018)
Women with endometriosis appear to be in a hypercoagulable state as manifested by shortened thrombin time (TT) and activated partial thromboplastin time (aPTT) but similar prothrombin time (PT), platelet count, and international normalized ratio (INR) to those without endometriosis(Ding, Liu et al. 2018). The TT (thrombin time) is involved in the conversion of fibrinogen to fibrin (Ding, Liu et al. 2018)
This suggests the activation of the intrinsic, but not extrinsic, coagulation pathway (Ding, Liu et al. 2018).
In addition, women with endometriosis had increased plasma levels of fibrinogen and platelet activation rate (Ding, Liu et al. 2018)
This newly found hypercoagulability provides a biologically plausible link between endometriosis and elevated risk of cardiovascular diseases and raises the possibility that these coagulation parameters could be used as biomarkers of endometriosis(Ding, Liu et al. 2018)
Compared with women without endometriosis, women with OMA (ovarian endometriomas) had a significantly higher platelet activation rate and platelet aggregation rate, elevated plasma D-dimer, fibrinogen, FDPs (fibrin degradation products), sP-sel (plasma soluble P-selectin), and F1þ2 (prothrombin fragment) levels as well as shortened TT (thrombin time). Interesting, 3 months after surgical removal of endometriotic lesions, TT (thrombin time) was prolonged and all the other coagulation measurements, except plasma fibrinogen level, were significantly reduced. These data, taken together, provide further evidence for hypercoagulability in women with endometriosis (Ding, Liu et al. 2018)
When blood clots and fibrin nets dissolve, FDPs (fibrin degradation products) are released into the circulation. If the system cannot dissolve blood clots, one may experience elevated FDPs. D-dimer is one specific FDP after the interlinking offibrin monomer and factor XIII (FXIII). When present in peripheral blood, D-dimer indicates the simultaneous activation of coagulation and fibrinolysis pathways (Ding, Liu et al. 2018)
Therefore, increased FDPs and D-dimer levels reflect the activation of coagulation and the fibrinolytic system and as such can be used as indicators of coagulation activation(Ding, Liu et al. 2018).
Therefore, the evidence of a hypercoagulable state in women with endometriosis includes: shortened TT, increased platelet activation rate and platelet aggregation rate, and elevated plasma D-dimer, fibrinogen, FDPs, sP-sel, and F1þ2. As such, this provides a biologically plausible link between endometriosis and elevated risk of cardiovascular diseases (Ding, Liu et al. 2018).
Polycystic Ovarian Syndrome
Several studies of women with PCOS have shown dysregulation of the hemostatic system (Mannerås-Holm, Baghaei et al. 2011). PCOS is characterized by a prothrombotic state, as reflected by increased PAI-1 activity and fibrinogen, without signs of dyslipidemia or a proinflammatory state (Mannerås-Holm, Baghaei et al. 2011). Polycystic ovary (PCO) syndrome (PCOS), the most common endocrine abnormality in premenopausal women, is characterized by hyperandrogenism, ovulatory dysfunction, and PCO morphology
Prothrombic state and PCOS
Women with PCOS are characterized by a prothrombotic state, as reflected by increased PAI-1 activity and fibrinogen (Mannerås-Holm, Baghaei et al. 2011). These disturbances are independent of BMI and are observed in the absence of hypertension, dyslipidemia, and low-grade inflammation(Mannerås-Holm, Baghaei et al. 2011). The elevated PAI-1 activity was associated with insulin resistance characterized by low SHBG and high insulin concentrations(Mannerås-Holm, Baghaei et al. 2011). On the other hand, inflammatory markers hs-CRP and sE-selectin together with HR best explained high fibrinogen levels in women with PCOS. SHBG and insulin were not associated with PAI-1 activity in controls, indicating that these factors have a specific impact in PCOS (Mannerås-Holm, Baghaei et al. 2011)..
Insulin resistance and hypercoagulability
Women with PCOS are prone to develop insulin resistance with associated hyperinsulinemia, dyslipidemia, hypertension, and obesity (Mannerås-Holm, Baghaei et al. 2011)..
These are all components of the metabolic syndrome that increase the risk for type 2 diabetes and cardiovascular (CV) events (Mannerås-Holm, Baghaei et al. 2011).
Low SHBG and high insulin levels were the strongest explanatory factors of high PAI-1 activity in women with PCOS(Mannerås-Holm, Baghaei et al. 2011). Hyperinsulinemia due to insulin resistance impairs fibrinolysis by enhancing PAI-1 secretion (Mannerås-Holm, Baghaei et al. 2011). Insulin also inhibits hepatic production of SHBG(Mannerås-Holm, Baghaei et al. 2011). Elevated insulin and low SHBG levels in women with PCOS partly explain the difference in PAI-1 activity between the groups because the significant difference in PAI-1 activity was lost after adjusting for BMI/age and SHBG/insulin. Thus, disturbed fibrinolysis in women with PCOS, indicated by high PAI-1 activity, is largely associated with variables closely related to insulin resistance(Mannerås-Holm, Baghaei et al. 2011).
Some results point to a prothrombotic state, including :
- Hypofibrinolysis
- hypercoagulability
- endothelial dysfunction
- platelet dysfunction
However, it unclear whether the hemostatic disturbances are independent of obesity (Mannerås-Holm, Baghaei et al. 2011). It is also unclear whether hyperandrogenemia, with high circulating testosterone and low SHBG, contributes to a disturbed fibrinolysis/coagulation in women with PCOS.
Women with PCOS have high circulating concentrations of PAI-1 activity and fibrinogen after adjustment for age and BMI(Mannerås-Holm, Baghaei et al. 2011).
PAI-1 is produced in adipocytes, endothelial cells, and hepatocytes in response to hormonal, metabolic, and inflammatory stimuli (Mannerås-Holm, Baghaei et al. 2011). PAI-1 is the principal inhibitor of fibrinolysis and the most extensively studied of the hemostatic factors in the metabolic syndrome and in PCOS(Mannerås-Holm, Baghaei et al. 2011).
Increased PAI-1 activity is associated with hypofibrinolysis and may contribute to thrombosis (Mannerås-Holm, Baghaei et al. 2011).
Fibrinogen
Elevated levels of fibrinogen, factor VIII, and vWF could lead to hypercoagulability. Fibrinogen is mainly produced by hepatocytes in response to inflammatory cytokines, and increased levels are associated with increased risk of CV events (Mannerås-Holm, Baghaei et al. 2011).
Fibrinogen may contribute to CVD by increasing fibrin formation, plasma viscosity, and platelet aggregation (Mannerås-Holm, Baghaei et al. 2011).
All phenotypic subgroups of PCOS appear to have higher fibrinogen levels than controls, but after adjustments for BMI and age only women with NIH-defined PCOS and normoandrogenic PCOS women were significantly different from controls. Thus, women with PCOS may have obesity-independent elevation of fibrinogen, which could cause hypercoagulability (Mannerås-Holm, Baghaei et al. 2011).
Lipid abnormalities may trigger platelet activation, activate the coagulation pathway, and inhibit fibrinolysis and are consequently associated with CVD risk (Mannerås-Holm, Baghaei et al. 2011).
Dyslipidemia
Several studies of dyslipidemia and PCOS have reported an atherogenic lipid profile, predominantly increased TG and decreased HDL-C levels (Mannerås-Holm, Baghaei et al. 2011).
We found higher TG levels in PCOS women than controls, but the difference was not significant after adjustments for age and BMI. TC, HDL-C, and LDL-C were similar in the groups. Nevertheless, all lipid variables correlated with fibrinogen and PAI-1 activity in women with PCOS.
Inflammation
Elevated levels of inflammatory mediators in women with PCOS have suggested that PCOS is characterized by low-grade chronic inflammation (Mannerås-Holm, Baghaei et al. 2011).
Inflammatory markers are predictors of CVD, and the levels of most hemostatic factors correlate with inflammation, as determined by hs-CRP (Mannerås-Holm, Baghaei et al. 2011).
Hence, low-grade chronic inflammation may increase CVD risk in PCOS by disturbing coagulation and fibrinolysis (Mannerås-Holm, Baghaei et al. 2011).
hs-CRP was increased in women with PCOS, but the difference was not significant after adjustments for age and BMI, indicating that an inflammatory state is due to obesity(Mannerås-Holm, Baghaei et al. 2011).
Other inflammatory markers that were investigated (IL-6, TNF-a, sICAM-1, sVCAM-1, and sE-selectin), did not differ in women with PCOS and controls, although several correlated positively with fibrinogen and PAI-1 activity in PCOS women.
I recently learned from Ruth Kriz that there are some genetics and pathways associated with hypercoagulation as well.
These include:
PAI-1 (plasminogen activator inhibitor) mutation– 4G deletion (5G/5G is normal)- just one copy will impair ability to convert plasminogen to plasmin (questions I ask: family history of CV disease, history of chronic infections, ear infections, sinus infections, men have prostatitis)- This population generally does not respond well to antibiotics.
Patients with the 4G/4G and 4G/5G deletion have the potential to form deleterious biofilms!
Factor V Leiden Mutation- factor V Leiden (FVL) is a point mutation of factor V resulting in an elimination of the cleavage site in factor V and factor Va. This genetic defect increases the risk of thrombosis, especially in homozygous or pseudo-homozygous FVL-mutated individuals. However, many individuals with the mutation may never develop a venous thrombotic event (VTE).
Thrombin activates factor V, and once activated, it will convert prothrombin to thrombin. Activated protein C, one of the principal physiologic inhibitors of coagulation, degrades factor V. In the presence of what is called thrombomodulin, thrombin acts to decrease clotting by activating protein C; therefore, the concentration and the action of protein C are important determinants in the negative feedback loop through which thrombin limits its activation.
Factor V Leiden is an autosomal dominant genetic condition that exhibits incomplete penetrance, meaning that not every person with the mutation will develop the disease.
Lp(a)- This is often associated with cardiovascular disease. There are some conflicts in the literature about the role it plays, and some say it can be protective. However, one thing we know: Lp(a) binds up TPA -Tissue plasminogen activator-(TpA) is made by endothelial tissue-that makes plasminogen. Without plasminogen, the blood clotting risk increases.
NO2 and platelet activation
Nitric oxide (NO) is an important cellular signaling molecule that participates in diverse physiological functions in mammals, including vasodilation, smooth muscle relaxation, neurotransmission, and the immune response (Cinelli, Do et al. 2020).
Nitric oxide (NO) exerts important vasodilatory, antiplatelet, antioxidant, antiadhesive, and antiproliferative effects(Gkaliagkousi, Ritter et al. 2007).
NO, a free radical, is produced by a family of enzymes called nitric oxide synthases (NOSs) by the oxidation of L-arginine (L-Arg) to L-citrulline(Cinelli, Do et al. 2020)
Nitric oxide (NO) is formed from the amino acid L-arginine by a 2-step oxidation of L-arginine to L-citrulline catalyzed by the enzyme nitric oxide synthase (NOS) (Gkaliagkousi, Ritter et al. 2007).
There are three isoforms of NOS. Two of them, neuronal NOS (nNOS) and endothelial NOS (eNOS), are constitutively expressed, while the third one is inducible and is thus termed iNOS (Cinelli, Do et al. 2020)
Of the 3 isoforms identified to date, NOS3 (endothelial-type NOS, NOS III) is expressed constitutively in endothelial cells and is the main source of vascular NO under physiological conditions, although small amounts of NOS2 (inducible NOS, NOS II) have also been reported to be expressed in the endothelium (Gkaliagkousi, Ritter et al. 2007)
By inducing vasodilation, endothelium-derived NO contributes to the maintenance of basal vascular tone and blood flow, and thus to the physiological regulation of blood pressure(Gkaliagkousi, Ritter et al. 2007); it also has important antiplatelet, antioxidant, antiadhesive, and antiproliferative properties.
Other cells expressing NOS3 in the cardiovascular system include cardiac myocytes,9 red blood cells, megakaryocytes, and platelets (Gkaliagkousi, Ritter et al. 2007)
iNOS and inflammation
The primary role of iNOS in human physiology is the destruction of invading pathogens(Cinelli, Do et al. 2020).
iNOS and NO are produced as a part of the immune response and have a regulatory role(Cinelli, Do et al. 2020).
NO’s high affinity for iron allows it to break up or inactivate heme-containing enzymes or iron-sulfur clusters(Cinelli, Do et al. 2020).
NO can be responsible for cysteine nitrosylationor can combine with superoxide to form ONOO– (peroxynitrite), a potent oxidant and nitrating agent. NO and ONOO– can cause DNA damage via oxidative deamination (Cinelli, Do et al. 2020).
The short biological half-lives of NO and ONOO– direct these damaging mechanisms preferentially to the invading pathogen, thus avoiding systemic toxicity(Cinelli, Do et al. 2020).
Finally, cyclic guanosine monophosphate (cGMP) production is increased through NO mediated activation of guanylate cyclase. cGMP is a soluble second messenger that participates in many downstream signaling cascades through activation of various protein kinases, ion channels, and phosphodiesterases(Cinelli, Do et al. 2020).
A considerable number of human diseases have an inflammatory component, and a key mediator of immune activation and inflammation is inducible nitric oxide synthase (iNOS), which produces nitric oxide (NO) from L-arginine (Cinelli, Do et al. 2020)
Inappropriately high NO concentrations from overexpression or dysregulation of iNOS, on the other hand, can result in toxic effects and is associated with a variety of human diseases, including septic shock, cardiac dysfunction, pain, diabetes, and cancers (Cinelli, Do et al. 2020)
NOS induction may also be linked to diabetes and obesity-associated insulin resistance, as it is known that iNOS is expressed throughout insulin-sensitive tissues, and obesity increases iNOS levels in muscle, adipose tissue, and vasculature (Cinelli, Do et al. 2020)
Studies in rat models show that rat pancreatic islet cells are very susceptible to NO toxicity and iNOS overexpression is found in pancreatic macrophages of prediabetic rats (Cinelli, Do et al. 2020)
iNOS’ role in insulin resistance may in part be through S-nitrosylation or tyrosine nitration of insulin signaling protein (Cinelli, Do et al. 2020)
Many potent iNOS inhibitors with high selectivity over related NOS isoforms, neuronal NOS (nNOS) and endothelial NOS (eNOS), have been discovered, and these drugs have shown promise in animal models of endotoxemia, inflammatory and neuropathic pain, arthritis, and other disorders(Cinelli, Do et al. 2020).
High levels of iNOS expression are also found in many cancerous tumors, including, colon, bladder, breast, and lung cancer (Cinelli, Do et al. 2020)
Various studies have also indicated that iNOS (both centrally and peripherally expressed) may play a role in the development and sensation of inflammatory and neuropathic pain(Cinelli, Do et al. 2020)
Meller et al. found that LPS and cytokines induce thermal hyperalgesia (hypersensitivity to pain), possibly mediated via induction of iNOS in spinal cord glial cells (Cinelli, Do et al. 2020)
Similarly, in animal models of neuropathic pain, iNOS upregulation contributes to local inflammatory reactions (Cinelli, Do et al. 2020)
Recently, even cancer-associated pain was found to be associated with upregulated iNOS (and nNOS) in mice with bone cancer (Cinelli, Do et al. 2020).
As such, treatment of pain has also been a major driving force for development of iNOS inhibitory therapeutics.
TNF, IL-1β, IFN-γ, and LPS are the main inducers of iNOS expression(Cinelli, Do et al. 2020)
Combinations of these inducers can generate synergistic effects in certain cells and are sometimes actually required to stimulate NO production, especially in nonimmune cells, where LPS only weakly induces iNOS (Cinelli, Do et al. 2020)
For instance, in mouse skeletal myocytes, none of these inducers alone can induce iNOS expression. However, stimulation of cells with both IFN-γ and TNF or IL-1 (or with all three cytokines) results in significant NO production (Cinelli, Do et al. 2020)
The stimuli and conditions that determine iNOS expression depend on the cell type and species. For example, LPS alone can stimulate iNOS expression in rat and mouse glial cells but not human ones (Cinelli, Do et al. 2020)
In addition to LPS and cytokines, invading viruses can also trigger iNOS induction in cells(Cinelli, Do et al. 2020)
Human immunodeficiency virus (HIV) induces iNOS expression in human glial cells, and high iNOS expression is also detected in hepatocytes of patients infected with hepatitis B virus (HBV). Transfection of hepatocytes with HBV genes results in iNOS expression, which indicates the virus can upregulate iNOS expression in human liver cells(Cinelli, Do et al. 2020).
iNOS upregulation genetics
Within my patient population, I have seen quite a bit of patients that have a phenotype associated with a polymorphism of iNOS, or NOS2, many with an upregulation. This can increase your risk of activated platelets, especially when there is weakness in pro-resolving mediators. An increase in activated platelets can increase formation of fibrin.
- Activated platelets secrete coagulation factors, expose PS, and support thrombin and fibrin formation (Swieringa, Spronk et al. 2018)
- Platelet receptors for collagen and thrombin are complementary and enforce each other’s activity(Swieringa, Spronk et al. 2018)
- The extrinsic and intrinsic pathways are inversed balanced in the formation of a platelet‐fibrin thrombus(Swieringa, Spronk et al. 2018)
- Clinical studies support the high degree of interactions between platelets and coagulation(Swieringa, Spronk et al. 2018)
Nitric oxide has different effects inside the body; it plays roles in platelet aggregation, cytotoxicity, blood flow, synaptic transmission, and neurotransmitters. Inducible nitric oxide, or iNOS, plays a stimulatory role in platelet activation. Platelets generate nitric oxide (NO) in response to agonist stimulation. In particular, there is an iNOS dependent nitric oxide production in platelets induced by thrombin.
Homocysteine can also increase platelet activation.
Homocysteine (Hcy) is a non-structural amino acid (AA) that is formed in the catabolic pathway for the essential amino acid, methionine (Met) (Morris, Kožich et al. 2017)
Met is converted to Hcy via S-adenosylmethionine (SAM) and Sadenosylhomocysteine (SAH), releasing a methyl group which is used in numerous methylation reactions (Morris, Kožich et al. 2017).
Hcy can be converted back to Met by the remethylation pathway. The methyl-group donor can either be 5-methyltetrahydrofolate (catalyzed by methionine synthase, with methylcobalamin as a cofactor) or betaine (Morris, Kožich et al. 2017).
Alternatively, Hcy is irreversibly metabolized to cysteine by the transsulfuration pathway. This starts with condensation of Hcy and serine to form cystathionine, catalyzed by CBS (Morris, Kožich et al. 2017).. Cystathionine is subsequently cleaved by cystathionine γ-lyase to form cysteine and 2- oxobutyrate. Cysteine can be further converted to taurine or to inorganic sulfate via hydrogen sulfide (Morris, Kožich et al. 2017).
In CBS deficiency, diminished cysteine production via the transsulfuration pathway may also contribute to low cysteine concentrations(Morris, Kožich et al. 2017).
CBS deficiency and homocysteine
CBS is expressed predominantly in liver, pancreas, kidney and brain. Its catalytic domain binds heme and the cofactor pyridoxal 5′phosphate, in addition to its substrates; the regulatory domain binds SAM as an allosteric activator
Hcy contains a thiol (–SH) group that reacts readily with other thiol groups. Hcy is, therefore, present in plasma in a variety of chemical forms.
Cystathionine beta-synthase (CBS) deficiency is a rare inherited disorder in the methionine catabolic pathway, in which the impaired synthesis of cystathionine leads to accumulation of homocysteine (Morris, Kožich et al. 2017).
The sum of the free and all bound Hcy species is defined as total homocysteine (tHcy). Cysteine also contains a thiol group and is present in different reduced and oxidized forms including mixed disulfides and protein-bound species. The binding capacity of plasma proteins is limited. As a result, grossly elevated concentrations of homocysteine lead to a decrease in plasma total cysteine(Morris, Kožich et al. 2017).
In CBS deficiency, diminished cysteine production via the transsulfuration pathway may also contribute to low cysteine concentrations(Morris, Kožich et al. 2017).
CBS deficiency homocysteine
Cystathionine beta-synthase (CBS) deficiency is a rare inherited disorder in the methionine catabolic pathway, in which the impaired synthesis of cystathionine leads to accumulation of homocysteine (Morris, Kožich et al. 2017).
Patients with CBS deficiency vary markedly in their symptoms, age of onset and rate of progression of clinical signs (Morris, Kožich et al. 2017). Pyridoxine-responsive patients generally have a milder phenotype and a later onset than the pyridoxine unresponsive ones (Morris, Kožich et al. 2017)
CBS is expressed predominantly in liver, pancreas, kidney and brain. Its catalytic domain binds heme and the cofactor pyridoxal 5′phosphate, in addition to its substrates; the regulatory domain binds SAM as an allosteric activator (Morris, Kožich et al. 2017).
CBS deficiency impairs the conversion of Hcy to cystathionine and leads to its accumulation (Morris, Kožich et al. 2017).
The pathophysiology of CBS deficiency is not fully understood. As well as the accumulation of Hcy, the defect leads to increased concentrations of SAH, enhanced remethylation to methionine, and depletion of cystathionine and cysteine (Morris, Kožich et al. 2017).
Raised Hcy concentrations modify sulfhydryl groups on proteins and interfere with the cross-linking of sulfhydryl groups in proteins such as elastin; this is thought to cause the lens dislocation and skeletal abnormalities(Morris, Kožich et al. 2017).
Raised Hcy concentrations also alter intracellular signaling and cause endoplasmic reticulum stress with endothelial dysfunction (Morris, Kožich et al. 2017).
These mechanisms together with impaired thrombolysis may be responsible for thromboembolism and vascular disease (Morris, Kožich et al. 2017).
Increased SAH impairs methylation reactions and decreased cystathionine and cysteine are associated with apoptosis, oxidative stress and alterations of structural proteins such as fibrillin, which may contribute to connective tissue abnormalities(Morris, Kožich et al. 2017).
Altered synthesis of hydrogen sulfide may also contribute to the pathophysiology(Morris, Kožich et al. 2017).
Patients with CBS deficiency show a wide spectrum of severity and age at presentation. Some patients have a severe childhood-onset multisystem disease, whilst others are asymptomatic into adulthood(Morris, Kožich et al. 2017). The main clinical features are dislocation of the optic lenses, osteoporosis and a ‘marfanoid’ habitus, learning difficulties and a predisposition to thromboembolism(Morris, Kožich et al. 2017)
Other causes of hyperhomocysteinemia include inborn errors of Hcy remethylation, vitamin deficiencies (especially B12), renal insufficiency and medication. (Morris, Kožich et al. 2017).
Mildly affected patients are likely to present as adults with thromboembolism and to respond to treatment with pyridoxine (Morris, Kožich et al. 2017).
Subsequently, connective tissue, neuro-psychiatric and vascular presentations have been recognized (Morris, Kožich et al. 2017)
Skeleton: patients tend to have excessive height and limb length (dolichostenomelia and arachnodactyly) and are prone to osteoporosis, at least after childhood. Various bone deformities and abnormal X-ray findings may also be present (Morris, Kožich et al. 2017).
Central nervous system: intellectual, psychiatric and behavioral problems are all common, especially in pyridoxine unresponsive patients (Morris, Kožich et al. 2017). Without treatment, approaching 90 % pyridoxine unresponsive patients have learning difficulties(Morris, Kožich et al. 2017). Seizures and extrapyramidal signs are also quite frequent features (Morris, Kožich et al. 2017).
Neurological complications include seizures and movement disorders in addition to strokes. Seizures occur in 20 % of untreated pyridoxine-unresponsive patients by 12 years of age(Morris, Kožich et al. 2017). EEG abnormalities may be more common, reflecting underlying brain disease. There is no evidence to support routine EEG monitoring in the absence of symptoms (Morris, Kožich et al. 2017).
There are several case reports of movement disorders which are not due to basal ganglia infarction, including polymyoclonus, dystonia and parkinsonism(Morris, Kožich et al. 2017).. Some of these patients have responded to L-DOPA therapy but others have not and this drug should be used with caution as it can raise Hcy levels
Vascular system: thromboembolism is the major cause of morbidity and early death. Venous thrombosis is more common than arterial but it can affect any part of the body (Morris, Kožich et al. 2017).
Homozygous cystathione β synthetase deficiency, causing homocystinuria, is associated with a thromboembolic event in 30% of patients by the age of 20 years, 50% of patients by the age of 30 years, and 60% of patients by the age of 40 years. Mild and moderately elevated HCY levels (hyperhomocysteinemia) are often seen(Morris, Kožich et al. 2017).
Vascular complications are common in untreated or poorly controlled CBS deficiency and include venous thrombosis and arterial thrombosis. Venous thrombosis is the more common.
A further meta-analysis showed that a 5 μmol/L elevation in HCY was associated with a 27% higher risk of venous thrombosis in prospective studies and 60% higher risk of venous thrombosis in retrospective studies. Genotype studies of the methylenetetrahydrofolate reductase (MTHFR) enzyme showed an association of venous thrombosis with hyperhomocysteinemia (Den Heijer, Lewington et al. 2005).
In 1964, McDonald et al concluded that homocysteine, both in vivo and in vitro, increased platelet stickiness associated with arterial and venous thrombosis, but this finding was not confirmed(Den Heijer, Lewington et al. 2005)
In studies of platelet kinetics, a direct relationship was found between HCY concentration and platelet survival. Harker et al found that increased platelet activation(Den Heijer, Lewington et al. 2005).
It has been suggested that the risk of thrombosis in homocystinuria depends largely on whether patients also have the factor V Leiden mutation (Morris, Kožich et al. 2017).
In patients with CBS deficiency, lowering the plasma tHcy is the single most important factor in reducing the risk of thromboembolic disease(Morris, Kožich et al. 2017).
There is some evidence that there may be increased platelet activation in patients with CBS deficiency, even on treatment (Morris, Kožich et al. 2017).
Acute stroke can occur in CBS deficiency patients due to arterial thrombosis/embolism and carotid artery disease/ dissection but it is usually due to cerebral venous sinus thrombosis (Morris, Kožich et al. 2017).
Severely affected patients with CBS deficiency share some clinical features with Marfan syndrome, including tall slender body habitus, arachnodactyly, lens dislocation and myopia (Morris, Kožich et al. 2017).
Other genetic conditions associated with non-traumatic lens dislocation include Weill Marchesani syndrome, Ehlers Danlos syndrome and sulfite oxidase deficiency (SUOX) (Morris, Kožich et al. 2017).
To ensure that a treatable diagnosis is not missed, CBS deficiency should be excluded in all cases by measuring plasma tHcy (Morris, Kožich et al. 2017)..
CBS deficiency is characterized biochemically by an accumulation of Hcy, decreased synthesis of cystathionine and cysteine and usually by increased Met. However, the classical pattern is not always observed in all variants of CBS deficiency (Morris, Kožich et al. 2017)..
In untreated patients with CBS deficiency, plasma tHcy exceeds the upper limit of the reference range (which is generally between 10 and 15 μmol/L, though it varies with age and method of analysis); tHcy concentrations are typically above 100 μmol/L but considerably lower concentrations may be found in patients with mild variants of CBS deficiency (Morris, Kožich et al. 2017)..
The median tHcy in a cohort of 25 CBS deficient patients was 125 μmol/L with a range of 16-281(Morris, Kožich et al. 2017).
Indeed, plasma tHcy concentrations may be normal in pyridoxine-responsive patients if they are receiving even low doses of pyridoxine in vitamins and food supplements (Morris, Kožich et al. 2017)..
Patients with CBS deficiency have low to low normal cystathionine and high to high normal methionine concentrations (Morris, Kožich et al. 2017).
In contrast, genetic and nutritional disorders of Hcy remethylation lead to raised plasma cystathionine and low or low normal methionine concentrations (Morris, Kožich et al. 2017)..
Other causes of hyperhomocystinaemia include renal failure, nutritional vitamin B12 and folate deficiencies and genetic disorders of vitamin B12 absorption or the Hcy remethylation pathway(Morris, Kožich et al. 2017).. Decreased or low normal plasma Met, elevated plasma cystathionine and/or elevated plasma or urinary methylmalonic acid concentrations suggest causes of hyperhomocysteinemia other than CBS deficiency (Morris, Kožich et al. 2017).
Folate deficiency is particularly likely to cause elevated tHcy concentrations in subjects who are homozygous for the common c.677C > T variant in the MTHFR gene(Morris, Kožich et al. 2017).
The patient’s history is also important as it may reveal other diseases associated with hyperhomocysteinemia or the administration of drugs such as nitrous oxide, methotrexate and other folate antagonists (Morris, Kožich et al. 2017)..
Dietary management of high homocysteine
Restricting intake of the essential amino acid, Met, reduces the precursor load on the transsulfuration pathway, thereby reducing Hcy production(Morris, Kožich et al. 2017). In most pyridoxineunresponsive patients, the biochemical targets can only be achieved by a diet that is very low in natural protein, with supplements of a Met-free L-AA mixture (Morris, Kožich et al. 2017).
Additional treatment with betaine can help patients who find it difficult to adhere to dietary restrictions and to attain good metabolic control (see Betaine treatment). Betaine lowers Hcy levels, potentially allowing an increase in Met intake (Morris, Kožich et al. 2017).
The level of Met or natural protein restriction required varies and is determined for each patient according to their plasma tHcy and Met concentrations(Morris, Kožich et al. 2017).
Other treatment recommendations include the use of betaine, especially for patients who have a difficult time adhering to the low methionine diet. Betaine (N,N,N-trimethylglycine) is formed in the body from choline and small amounts are present in the normal diet. lowers Hcy concentrations in CBS deficiency by donating a methyl group and converting Hcy to Met (Morris, Kožich et al. 2017).
Betaine may also act as a chemical chaperone and correct partial mis-folding of CBS mutants (Morris, Kožich et al. 2017).
Individuals with plasma Met concentrations greater than 80 μmol/L respond less well to betaine (Morris, Kožich et al. 2017).
Betaine is generally safe but some people dislike the taste and compliance may be poor (Morris, Kožich et al. 2017). It can result in a fishy odor (Morris, Kožich et al. 2017). This is probably due to inadequate activity of flavin containing monooxygenase 3 and may respond to riboflavin (Morris, Kožich et al. 2017).
Nutritional vitamin B12 and folate supplementation can also be helpful for patients with mutations in Hcy remethylation pathway or B12 absorption issues (such as with GI disorders).
MTHFR
Many patients have polymorphisms in an enzyme that increases the risk of high homocysteine- MTHFR C677T. The 5,10-methylenetetrahydrofolate reductase (MTHFR) gene polymorphism C677T has been shown to result in increased total homocysteine concentrations on the basis of low folate levels caused by decreased enzyme activity.
A common polymorphism exists in the gene that encodes the catalytic domain of the MTHFR enzyme, in which a C > T substitution at position 677 (referred to as 677TT) results in a substitution of alanine to valine (Den Heijer, Lewington et al. 2005)
The single amino acid substitution results in impaired folate binding and reduced activity of the MTHFR enzyme (Den Heijer, Lewington et al. 2005)
About 10–12% of Caucasians of Northern European descent carry the 677TT genotype for MTHFR and have about 25% higher homocysteine levels than those with the 677CC genotype(Den Heijer, Lewington et al. 2005)
The effect of the MTHFR 677TT genotype on homocysteine levels varies according to folate status, whereby the difference in homocysteine levels between those with TT vs. CC genotypes is greater among those with low folate or riboflavin status than in those with normal or high levels of these vitamins (Den Heijer, Lewington et al. 2005)
A meta-analysis published in 2005 (Den Heijer et al) demonstrated that the 677TT genotype was associated with a 20% increased risk of venous thrombosis (Den Heijer, Lewington et al. 2005)
A meta-analysis of the short-term trials of folic acid-based vitamin supplements showed that a daily dose of folic acid of between 0.5 and 5 mg was associated with a reduction in homocysteine levels of 25% and that vitamin B12 was associated with a further reduction of about 7% (Den Heijer, Lewington et al. 2005)
This difference in homocysteine of 25% that is typically achieved with folic acid supplements is comparable to the difference in homocysteine between 677TT and 677CC genotype (Den Heijer, Lewington et al. 2005)
In clinical practice, measurement of homocysteine levels (and subsequent determination of folate and vitamin B12 levels if homocysteine levels are elevated) may be indicated for individuals with unexplained idiopathic venous thrombosis or recurrent venous thrombosis, or venous thrombosis occurring at early age or at an unusual site (Den Heijer, Lewington et al. 2005)
Vitamin supplements may be indicated for those individuals who are identified with evidence of folate or vitamin B12 deficiency [98], but whether such supplements may reduce the risk of recurrent venous thrombosis is not known (Den Heijer, Lewington et al. 2005)
Therapeutic strategies
SPM’s to the rescue
Specialized pro-resolving mediators (SPMs) are lipid mediators derived from poly-unsaturated fatty acids (PUFAs). They have shown to have an important role in the inflammation environment, preventing an overreaction of the organism and promoting the resolution of inflammation(Valente, Dentoni et al. 2022).
SPMs are lipid mediators (LMs) derived from PUFAs (poly-unsaturated fatty acids), such as AA (Arachidonic Acid), EPA (eicosapentaenoic acid), DHA (docosahexaenoic acid) and n-3 DPA (n-3 docosapentaenoic acid).
SPMs include lipoxins, resolvins, protectins and maresins, as well as newly identified cysteinyl-conjugated SPMs (cys-SPMs) and n-3 DPA-derived SPMs.
Brief description of these components are below:
Lipoxins– the first discovered SPM, derived from eicosanoids derived from arachidonic acid. Aspirin can trigger their biosynthesis.
Resolvins- Resolvins are generated in inflammatory exudates during the phase of resolution. They derive from ω-3 fatty acids EPA and DHA, forming E series (RvE) and D series (RvD) resolvins, respectively. Their synthesis is also promoted by aspirin (likewise Lipoxins). They are known to inhibit the production of pro-inflammatory mediators , enhance scavenging of pro inflammatory cytokines and promote recruitment of monocytes and phagocytic clearance via lymphatic system.
Protectins- Protectins consist of Protectin D1/Neuroprotectin D1 (PD1/NPD1). They are biosynthesized from DHA via the 15-LOX mechanism. It can be found in human cell types, murine exudates and brain tissue, PD1/NPD1 has neuroprotective properties in the brain, retina and Central Nervous System (CNS). Its aspirin-triggered epimer, 17R-NPD1, has the same actions as NPD1 in controlling PMN, enhancing macrophage functions and attenuating experimental stroke.
Maresins
Maresins were first identified in human macrophages, in a pathway initiated by 12-LOX. Their name derives from an acronym: Macrophage Mediators in Resolving Inflammation. Maresin1 (MaR1) is able to promote the regeneration of tissues in an experimental model of simple organisms (planaria) with a strong capability of regeneration. In human cells, it is produced by platelets and PMN interactions. MaR1 promotes tissue regeneration and repair and has a neuroprotective role.
It is important to emphasize that these endogenous mediators of resolution do not act to “inhibit” of inflammation pathways: instead, they actively promote specific pathways in order to obtain a return to homeostasis.
How it works
The first signs of inflammation response are vasodilation and changes in vessel permeability. These factors not only permit the recruitment of cells implied in the inflammatory response but also give substrates for the biosynthesis of important molecules, such as SPMs. Apparently, ω-3 PUFAs, AA, EPA and DHA can be found within inflammatory exudates during very early phases, as demonstrated in various works. Therefore, the inflammation response is counterbalanced early by pro-resolution mediators. This avoids an excess of an inflammatory response that can be disruptive for the organism and for the tissue itself.
From the beginning of the inflammation process, SPMs reach the site of edema, either transported by blood flow or produced within the inflammatory tissue.
What SPMs do is signal the immune system to stop actively responding to pro-inflammatory signals, and instead to accelerate the return to homeostasis. SPMs play a unique role in helping the body to shut down the immune response, inhibit additional inflammation, clear away the damaging byproducts of inflammation, and aid in tissue remodeling.
SPMs can facilitate the resolution even of prolonged or chronic inflammation. And once the SPMs have done their job, the body naturally breaks them down and eliminates them.
Obstacles
Optimal SPM production is highly dependent on both genetic predispositions and on good nutrition and a healthy lifestyle. Factors that can limit SPMs production include:
- Overload of environmental irritants
- High intake of low-quality dietary fats
- High intake of processed carbohydrates
- Too little exercise or overexertion
- Physical stressors
- Insufficient or poor-quality sleep
- Aging
Many people either have mutations in FAD’s, or they do not eat enough fatty fish. This is necessary to increase the pool of SPM’s. Main receptors for specialized pro-resolving lipid mediators (SPMs) in immune cells and platelets. The key effects of SPMs in immune cells are to decrease inflammation, chemotaxis and infiltration and increase phagocytic activity. SPMs also affect platelet function, decreasing aggregation, activation and clot remodeling.
SPMs reduce EC (endothelial cell) activation and proliferation, and migration and inflammation in VSMCs. SPMs also reduce immune cell activation and infiltration, promote macrophages M2 phenotype polarization, facilitate macrophage and PMN efferocytosis and inhibit platelet activation and aggregation.
Thus, in general, SPMs have beneficial antithrombotic properties, inhibit inflammatory responses induced by platelet-activating factor and avoid aggregation. These findings suggest that, in principle, any clinical condition that is characterized by immune cell infiltration or altered platelet reactivity might benefit from the effects of SPMs.
Vitamin K deficiency- a double edged sword.
As I mentioned earlier, vitamin K deficiency can impair the production of S protein- thus potentially leading to coagulation defects. However, it also can lead to hyperoxaluria and crystal deposition.
A study in 2019 in China demonstrated that VK1 (Vitamin K1) treatment can inhibit the formation of renal crystals in vivo. VK1 increases MGP Matrix GLA protein expression and functions through MGP to reduce crystal deposition in cells and provide cell protection. Their findings suggest that VK1 treatment could be a potential strategy for the treatment and prevention of nephrolithiasis. They found for the first time that VK1 plays a role in inhibiting crystal formation by decreasing crystal deposition and protecting cells from apoptosis and injury through MGP. Also, VK1 treatment may protect the kidney from inflammation and fibrosis.
VK is a liposoluble vitamin, including the naturally occurring VK1 and VK2 and the synthetic VK3 and VK4. VK1 is synthesized by plants and vegetables, whereas VK2 can be endogenously produced in animals or synthesized by intestinal bacteria. VK1 can be converted into VK2 and accumulated in extrahepatic tissues. The limited storage capacity of VK in the human body can easily lead to VK deficiency. Warfarin acts as a VK antagonist by blocking the VK cycle, resulting in functional VK deficiency. Extensive calcification was observed in the rat kidney after warfarin treatment.
The reason this is important is twofold. One, crystal formation can induce IL-6 which can indirectly activate the coagulation pathway via tissue factor as mentioned above. Second, vitamin K deficiency can impair the production of S protein, which is an anticoagulant protein.
Lumbrokinase
Earthworms have been used as a traditional medicine in China, Japan, and other Far East countries for thousands of years(Wang, Tull et al. 2013). Earthworms contain many compounds with potential medicinal properties and have been administrated to treat inflammatory, hematological, oxidative, and nerve disease.
Among many properties, earthworms also exhibit fibrinolytic activity. The pharyngeal region, crop, gizzard, clitellum, and intestine secrete an enzyme that plays a role in dissolving fibrin [9, 10] (Figure 1). Ground-up earthworm powder has been used as oral administration to support circulatory health and treat blood diseases.
In 1991, Dr. Mihara and other scientists in Japan successfully extracted and characterized a group of fibrinolytic enzymes from the earthworm species, Lumbricus rubellus (Wang, Tull et al. 2013). These enzymes are capable of degrading both plasminogen-rich and plasminogen-free fibrin. The enzymes were collectively named lumbrokinase (LK) after the genus name for earthworm, Lumbricus.
Oral administration of dry earthworm powder is considered as a potent and effective supplement for supporting healthy blood circulation(Wang, Tull et al. 2013). Lumbrokinases are a group of enzymes that were isolated and purified from different species of earthworms. These enzymes are recognized as fibrinolytic agents that can be used to treat various conditions associated with thrombosis.
Pharmaceutical thrombolytic agents typically used to dissolve clots are urokinase (u-PA), streptokinase, and tissue plasminogen activator (t-PA). These drugs, however, are not specific for fibrin and have adverse and dangerous side effects including severe bleeding and heavy blood loss which may result in death(Wang, Tull et al. 2013).
In contrast, LK is very specific to fibrin as a substrate and it does not cause excessive bleeding (Wang, Tull et al. 2013). It can dissolve the fibrin itself or convert plasminogen to plasmin by inducing endogenous tPA activity to dissolve fibrin clots .
LK has shown therapeutic promise for use in dissolving clots, lowering whole blood viscosity, and reducing platelet aggregation(Wang, Tull et al. 2013). It has not shown any adverse effects on the functions of the nervous system, respiratory system, cardiovascular vessels, or the liver and kidney (Wang, Tull et al. 2013). Currently, LKs are widely used clinically as a thrombolytic agent in China to treat cerebral infarction, coronary heart disease, pulmonary heart disease, deep vein thrombosis. angina pectoris, diabetes, and cerebral infarction. In Japan, Korea and also in North American countries such as Canada and the United States, LK has been used as oral supplement to support and maintain healthy cardiovascular function (Wang, Tull et al. 2013)
Royal Jelly
Royal jelly (RJ), a highly nutritious natural product, has gained recognition for its remarkable health-promoting properties, leading to its widespread use in the pharmaceutical, food, and cosmetic industries(Bagameri, Botezan et al. 2023)
Royal jelly (RJ) is a yellowish-white, creamy substance secreted by the hypopharyngeal and mandibular glands of worker bees. It serves as the primary nourishment for developing larvae during their initial three days and continues to be the exclusive food source for honeybee queens throughout their lives.
Recognized for its exceptional nutritional value, RJ has gained the reputation of a “superfood” with numerous potential health benefits for human consumption. It is characterized by its creamy texture and has a pH range of 3.6–4.2. It comprises a diverse range of essential components, including proteins, lipids, carbohydrates, minerals, amino acids, vitamins, enzymes, and hormones. These constituents contribute to its remarkable nutritional profile and may play a role in its physiological effects.
RJ’s rich composition not only holds potential for pharmaceutical applications but also for use in dietary supplements and functional foods, aiding in overall health and well-being. As the critical sustenance for queen bees, RJ necessitates the inclusion of all life-supporting nutrients, encompassing sugars, proteins, lipids, and water, harmoniously proportioned. RJ stands as the bee product with the most significant water content, accounting for 60–70%, while exhibiting a lower sugar ratio, 7–16%, relative to other bee derivatives. It also manifests a high protein content, spanning 10–18%, and lipids ranging between 3–8%. The composition extends to phenolic compounds, flavonoids, organic acids, minerals, vitamins, enzymes, and hormones.
In addition to these proteins and peptides, RJ also encompasses a collection of free amino acids, which includes lysine, proline, cystine, aspartic acid, valine, glutamic acid, serine, glycine, cysteine, threonine, alanine, tyrosine, phenylalanine, leucine, isoleucine, and glutamine.
RJ is recognized for its complex and diverse nutrient content, which encompasses an array of carbohydrates. These include glucose, fructose, sucrose, maltose, turanose, trehalose, and isomaltose(Bagameri, Botezan et al. 2023)..
RJ is a remarkable substance, rich in a range of lipids. Its lipid composition is predominantly composed of medium-chain fatty acids, among which 10-HDA is a major constituent. However, other fatty acids, such as sebacic acid and 9-hydroxy-2-decenoic acid, are also present and contribute to the overall lipid matrix of RJ. Beyond fatty acids, RJ also comprises other lipid-soluble substances, including waxes, sterols, and phospholipids(Bagameri, Botezan et al. 2023)..
RJ is characterized by its phenolic compounds, comprising substances such as pinobanksin, hesperetin, naringenin, isosakuranetin, chrysin, acacetin, luteolin, apigenin, kaempferol, isorhamnetin, formononetin, along with various glycosides. These compounds contribute to the biological activities and potential health benefits associated with RJ(Bagameri, Botezan et al. 2023).
The vitamin profile of RJ is also noteworthy. It includes essential vitamins, such as biotin, riboflavin, thiamin, pantothenic acid, inositol, niacin, pyridoxine, and vitamin E. These vitamins are key to multiple biological functions, further enhancing the nutritional value and health-promoting potential of RJ. This bee product also incorporates a range of minerals, such as potassium (K), phosphorus (P), calcium (Ca), magnesium (Mg), sodium (Na), zinc (Zn), chromium (Cr), and cadmium (Cd). The presence and concentration of these minerals in RJ are affected by a wide range of factors. These include environmental conditions, the time of the harvest season, and the unique biological traits of the bees. Therefore, these factors should be taken into account when studying the composition and properties of RJ
Extensive investigations have revealed that RJ possesses a broad spectrum of therapeutic effects, including anti-inflammatory, antioxidant, antitumor, anti-aging, and antibacterial activities (Bagameri, Botezan et al. 2023).
Royal jelly (RJ), a bee product produced by the hypopharyngeal and mandibular glands of worker bees, is chemically composed of water, proteins, carbohydrates, vitamins, lipids, and other bioactive compounds (You, Chen et al. 2018)
Moreover, RJ contains a diverse array of bioactive substances, such as lipids, phenolic compounds, flavonoids, organic acids, minerals, vitamins, enzymes, and hormones
. As a functional food, RJ has various pharmacological activities, including anti-inflammatory, antihypertensive, antioxidative, and memory function restoration activities (You, Chen et al. 2018)
A study by You et al (2018) demonstrated that RJ significantly inhibited iNOS and COX-2 expression at mRNA and protein levels. The mRNA expression of IL-6, IL-1β, and TNF-α was also downregulated by RJ in a concentration-dependent manner. Additionally, RJ protected BV-2 cells against oxidative stress by upregulating heme oxygenase-1 (HO-1) expression and by reducing reactive oxygen species (ROS) and nitric oxide (NO) production (You, Chen et al. 2018).
Numerous biological and chemical factors, including cytokines, pro-inflammatory enzymes, as well as the enzymatic degradation of tissues, all contribute to the inflammatory process. Interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) production are effectively inhibited by RJ treatment in a dose-dependent manner without having any deleterious effects on macrophages in vitro(You, Chen et al. 2018).
LPS can significantly increase NO production. It was reported that LPS stimulation induces inflammation by activating NF-κB and MAPK pathways. LPS treatment in BV-2 cells also caused oxidative stress by increasing ROS levels(Bagameri, Botezan et al. 2023)
The production of ROS could oxidize proteins, lipids, and DNA, and it is considered to be a vital mediator in conditions like AD. They observed that RJ pretreatment showed significant protective effects in BV-2 cells by scavenging ROS compared to the LPS-treated group(You, Chen et al. 2018).
This means that RJ could work well to maintain cellular redox balance and may inhibit the onset or progress of some oxidative-related diseases, such as neurodegenerative diseases.
HO-1 (hem oxygenase -1), an antioxidant gene, is related to immunologic defense in immune cells, including BV-2 microglia cells. Previous studies have reported that various compounds exert the anti-inflammatory activity in microglial cells not only through inhibiting NF-κB pathway, but also through activating Nrf2 pathway and antioxidant response element (ARE), as well as through increasing the synthesis of some antioxidant enzymes, including HO-1
RJ, according to their results, upregulated the mRNA expression of HO-1 and also exerted anti-inflammatory effects via NF-κB pathway. They speculate that the anti-inflammatory effects of RJ may be attributed to the regulatory effects of HO-1.
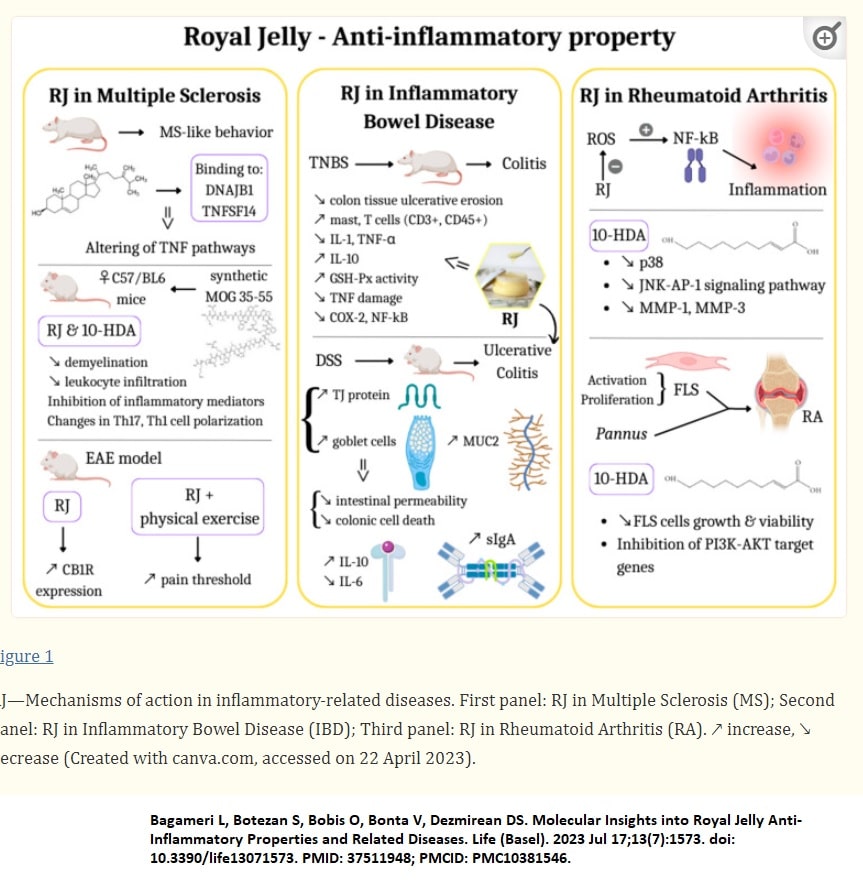
Phenolic compounds
As mentioned earlier, there are a range of hemostasis dysfunctions, such as coagulation dysfunctions and changes in blood platelet functions, that can be associated with many pathologies including death. These complications may be associated with oxidative stress.
A diet rich in fruits and vegetables has a significant influence on hemostasis and CVDs. Various authors have found that fruit and vegetable consumption influence the activity of various elements of hemostasis, such as coagulation proteins, and by reducing oxidative stress in the cardiovascular system and inhibiting blood platelet activation (Olas 2022)..
Phenolic compounds are not only found in medicinal plants, but also in the edible parts of the fruits and vegetables which form a key part of the human diet; they have also been identified in chocolate, dry legumes and cereals, and beverages such as tea, coffee and wine (Olas 2022).
Phenols can be divided into several groups based on their chemical structure, such as flavonoids, phenolic acids, stilbenes, tannins, coumarins, lignans and curcuminoids; they can also occur in free forms or conjugated with sugars and acids (Olas 2022).
Resveratrol
Resveratrol is a secondary metabolite produced by some species in response to harmful environmental factors, such as UV radiation, fungal, bacterial and viral infection, or mechanical damage to the plant.
The best and most widely available natural source of resveratrol is the grape (Vitis vinifera), especially the darker varieties (Olas 2022). However, resveratrol is also found in mulberry, black currant, blueberry, cowberry and American, strawberries, raspberries, apples and peanuts.
Resveratrol has also been found to inhibit platelet function (Olas 2022). This may be associated with partial modification of flux in the polyphosphoinositide cycle and a decrease in phosphatidylinositol 4,5-bisphosphate available for signaling in blood platelets (Olas 2022).
Inhibition of polyphosphoinositide metabolism may be associated with inhibition of PI-4(5)-kinase and protein kinase C (Olas 2022). In addition, resveratrol has been found to reduce cyclooxygenase activity and inhibit arachidonic acid metabolism in platelets in vitro (Olas 2022).
Gligorijevic et al. report that resveratrol may bind to fibrinogen in vitro and protect it from harmful oxidation (Olas 2022). Other results indicate that resveratrol may interact with thrombin, which is an important protein for platelet activation and the coagulation process(Olas 2022) Resveratrol also demonstrated both anticoagulant and antifibrinolytic potential in human umbilical vein endothelial cell (HUVECs) cell culture(Olas 2022).
Scientists suggest that resveratrol may effect primary hemostasis, including the suppression of blood platelet activation (especially platelet aggregation) and secondary hemostasis (modulating coagulation process by the inhibition of tissue factor/factor VIIa complex formation (Olas 2022).
Anti-platelet action of resveratrol probably involves the inhibition of cyclooxygenase activity, lowering nitric oxide concentration, decreasing cytoplasmic Ca2+ and blocking Ca2+ entry into blood platelets (Olas 2022). Results of Jang et al. suggest that the anti-platelet action of resveratrol is associated with the inhibition of NADPH oxidase—derived ROS production (Olas 2022)
Resveratrol analogues have also anti-platelet potential(Olas 2022). Pterostilbene (a dimethylether analogue of resveratrol) possesses high potency in the prevention of blood platelet activation in humans. For example, it inhibits platelet aggregation stimulated by collagen and reduces P-selectin exposition (Olas 2022). Revishankar et al. also observed that other resveratrol analogue—isorhapontigenin inhibits blood platelet activation (Olas 2022).
Curcumin
Another natural phenolic compound with antioxidant properties is curcumin, which has been used in natural medicine for thousands of years. It is a plant compound found in the rhizome extract of Curcuma longa L, and is commonly used as a spice known as turmeric. It is used to impart a strong yellow color to foods, most famously Indian curry, but also to a range of other foods, such as margarine, mustard, pasta or soft drinks (Olas 2022). In traditional Indian and Chinese medicine is was used for:
- quell excessive appetite.
- treat jaundice.
- treat diseases of the liver and digestive system, as well as colic, tooth and chest pain
- menstrual pain (Olas 2022).
It was also used as a means of supporting wound healing and treating skin discoloration(Olas 2022).
In modern natural medicine, curcumin is regarded as an antiseptic, anti-inflammatory, antioxidant, anti-platelet and anti-cancer agent (Olas 2022) It is recommended for treating eye infections, skin conditions such as burns, bites and acne, and diseases of the digestive system such as diarrhea, indigestion, hyperacidity, flatulence and ulcers. It has even been recommended as an antidepressant.
Curcumin has the capability to scavenge various intracellular small oxidative molecules; it can also up-regulate the expression of glutathione and reduce the production of ROS (Olas 2022).
Kolodziejczyk et al. describe the antioxidative potential of curcumin (12.5–50 μg/mL) in human blood platelets and plasma treated with ONOO− (in vitro), and Manikandan et al. indicate that curcumin modulates ROS production in myocardial ischemia in rats, probably by inhibition of xanthine dehydrogenase and xanthine oxidase.
Kim et al. report that curcumin and its derivatives demonstrate anticoagulant properties in vitro and in vivo; they appear to prolong prothrombin time and inhibit the generation of thrombin and activity of coagulation factor Xa in human plasma (Olas 2022). Moreover, curcumin demonstrated a stronger anticoagulant effect than its derivatives, indicating that the methoxy group in curcumin positively regulated its anticoagulant function (Olas 2022).
Madhyastha et al. found curcumin to facilitate fibrinolysis and cellular migration by modulating urokinase plasminogen activator expression (uPA) (Olas 2022).
Scientists identified molecular mechanisms involved in the inhibitory effects of curcumin on blood platelet activation is by activation of adenosine A2A receptor stimulated protein kinase A activation and phosphorylation of vasodilator-stimulated phosphoprotein (Olas 2022). Results of Chapman et al. also demonstrated that the major plasma metabolite of curcumin—tetrahydrocurcumin may modulate hemostasis: platelet aggregation and coagulation process(Olas 2022).
Quercetin
One of the most important phenolic compounds in the diet is quercetin (from the Latin quercetum—oak forest). It is an almost ubiquitous compound supplied to the body via a fruit and vegetable diet. Quercetin is most often found in the form of glycosides, i.e., sugar derivatives, e.g., rutin, although it can also occur in a free form. It is commonly found in tea, red wine, onions, apples and medicinal herbs, although its highest concentration has been noted in capers (234 mg per 100 g of edible portion). Its daily consumption is estimated at 25–50 mg. However, quercetin has a low bioavailability after oral administered.
The biological action of quercetin results from its high antioxidant activity and the ability to inhibit enzymes involved in the formation of the inflammatory process(Olas 2022).
Its antioxidative properties are based on various mechanisms:
- scavenging ROS, inter alia by inactivating produced oxygen radicals (hydroxyl radical, singlet oxygen and lipid radicals);
- limiting the production of ROS in cells by inhibiting the activity of enzymes involved in their formation (xanthine oxidase, membrane oxidase NAD(P)H, myeloperoxidase);
- chelation of transition metal ions (copper and iron), which prevents the formation of reactive hydroxyl radicals in cells, an important factor in the pathogenesis of many diseases;
- breaking the cascade of free radical reactions in enzymatic and non-enzymatic lipid peroxidation;
- possibly affecting the transmission of intracellular signals by protein kinases. Quercetin also exhibits significant heart-related benefits, such as inhibition of low density lipoprotein (LDL) oxidation and exerting a protective effect on NO. function under oxidative stress
- The compound has also been found to influence the oxidative profile in blood platelets of rats with hypothyroidism.
Mosawy et al. studied the effect of 6 mg/kg quercetin, administered daily for seven day appeared to inhibit the activation of platelets.
In addition, quercetin (6 mg/kg, daily for seven days) was found to reduce platelet aggregation and indicated that quercetin inhibits platelet adhesion to collagen and fibrinogen. Its action is correlated with inhibition of the activity of various kinases.
However, metabolites of quercetin can be responsible for the antiplatelet effect. For example, methycatechol formed by human microflora has a strong antiplatelet effect. Its mechanism of action is mainly based on the effect on intracellular calcium signaling.
In conclusion, the antioxidant, anti-platelet and anticoagulant properties of phenolic compounds have been well documented in a number of in vitro and in vivo studies, which have demonstrated they can play a positive role.
Dietary modification
Several lines of evidence indicate that the gut microbiota plays a role in systemic inflammation, metabolic syndrome, vascular dysfunction and atherosclerosis(Velasquez, Ramezani et al. 2016). Thus, investigators have explored the possibility of modulating gut microbes and their metabolites for health benefits(Velasquez, Ramezani et al. 2016). .However, such efforts have been hampered by the complexity of the interaction of microbiome with host genetics, host diet, and other unknown factors. The discovery of TMAO as a potential gut derived toxin with cardiovascular disease has energized such efforts. A number of therapeutic strategies have been tested from a broad spectrum of antibiotics to target-specific molecules with varying levels of success.
Assuming the microbiome is the source of TMAO, antibiotic treatment should reduce its circulating levels. Indeed, in health participants, suppression of intestinal microbiota with oral broad spectrum antibiotic reduced plasma TMAO levels after a phosphatidylcholine challenge (Velasquez, Ramezani et al. 2016). TMAO levels increased one month after stopping antibiotics(Velasquez, Ramezani et al. 2016). However, antibiotics are not an ideal treatment since it could have other unwanted consequences, and also chronic treatment is likely to lead to emergence of resistant bacteria (Velasquez, Ramezani et al. 2016).
In addition to a diet low in TMAO precursors, a diet low in saturated fat is beneficial. As indicated earlier, healthy diets rich in unsaturated fatty acids have been associated with lower postprandial circulating levels of LPS, closely associated with lower pro-inflammatory markers. Conversely, the consumption of high-energy or high saturated-fat diets has been associated with increased postprandial levels of LPS and increased circulating levels of pro-inflammatory markers (André, Laugerette et al. 2019). Therefore, a diet higher in polyunsaturated fats should be taken priority and the consumption of saturated fats should be limited.
Probiotics
Probiotics are live microorganisms that confer a health benefit on the host (Cantero, Guedes et al. 2022).
Mechanisms associated with the beneficial effects of probiotics include production of inhibitory substances such as bacteriocins, competition with pathogenic bacteria for nutrients, blocking of adhesion sites for pathogenic bacteria, degradation of toxins and the blocking of toxin receptors, immunomodulation, and fortification of the intestinal barrier (Cantero, Guedes et al. 2022)
In addition, numerous studies reported that probiotics exert preventive and or therapeutic effects on CVD by improving lipid and metabolism profiles (Cantero, Guedes et al. 2022)
Spore based probiotics
Recently it has been speculated gram positive, spore-forming probiotic strains may be a good alternative because the endospores that encapsulate the strains are highly resistant to stomach acid, potentially resulting in the delivery of more viable probiotics to the small intestine (McFarlin, Henning et al. 2017)
The study by McFarlin (2017) resulted in a finding of a 42% reduction in metabolic endotoxemia. Several of our exploratory biomarkers were either significantly reduced (IL-12p70, IL-1β, and ghrelin) or trended toward reduction (IL-6, IL-8, and MCP-1) with spore-based probiotic supplementation(McFarlin, Henning et al. 2017).. It is reasonable to speculate that the spore-based probiotic supplement may have exerted its effect by altering the gut microbial profile, altering intestinal permeability, or a combination of the two effects (McFarlin, Henning et al. 2017)
Given the pro-inflammatory actions of IL-1β, the observed reduction with probiotic supplementation was consistent with reductions in post-prandial systemic inflammation(McFarlin, Henning et al. 2017). It is speculated there are a variety of metabolic actions, the chief action in the present study is the ability to modulate the release of TNF-α or related inflammatory cytokines following antigenic challenge(McFarlin, Henning et al. 2017). In the case of the present study, reduced IL-12p70 with probiotic supplementation may reflect a reduction in systemic inflammatory capacity. In addition to the biomarkers that reached significance, we also found similar numerical trends for IL-6, IL-8, and MCP-1, which are all released by adipose tissues and commonly elevated in obese individuals(McFarlin, Henning et al. 2017)
Probiotics and TMAO
The body of evidence on the use of probiotics to reduce TMAO show that only a few strains have this specific benefit. Lactobacillus rhamnosus GG was the most efficient strain in reducing the plasma TMAO level in both humans and animals and based on the results (Cantero, Guedes et al. 2022). Qiu et al. investigated the potential TMAO lowering property of five different probiotics strains, and only Lactobacillus plantarum ZDY04 was able to significantly lower the plasma TMAO levels.
This was achieved through the remodeling of the gut microbiota, and not by affecting the expression of hepatic FMO3 and metabolizing choline, TMA, and TMAO.
imilarly, another study reported the TMAO-lowering potential of Enterobacter aerogenes ZDY01 in choline-fed mice; the effect was also attained through gut remodeling (El Hage, Al-Arawe et al. 2023)
It has been reported that probiotics are able to improve the integrity of the epithelial barrier and support the function of tight junctions which can inhibit the translocation of harmful metabolites, such as TMAO and LPS, from entering the peripheral circulation leading to a stable atherosclerotic plaque (El Hage, Al-Arawe et al. 2023)
SL-3 is a well-studied probiotic mixture containing eight different probiotic strains: Bifidobacterium breve, Bifidobacterium longum, Bifidobacterium infantis, Lactobacillus acidophilus, Lactobacillus plantarum, Lactobacillus paracasei, Lactobacillus bulgaricus, and Streptococcus thermophilus. Different studies have shown the beneficial effects of VSL-3 in different diseases, including ulcerative colitis liver disease, and Crohn’s disease
Probiotics are also able to reduce inflammation by increasing the number of Treg cells (El Hage, Al-Arawe et al. 2023)
Besides removing TMAO producing bacteria, use of selected bacteria to increase its metabolism has also been attempted. TMA could be depleted in vivo by bioconversion into a molecule with no undesirable properties. Brugere et al. proposed the use of archaeobiotics to prevent trimethylaminuria accumulation and cardiovascular disease. This is based on the concept of reducing TMA formation in the gut by converting it into an inert molecule, such as methane (Velasquez, Ramezani et al. 2016).
A number of methanogenic bacteria belonging to the order Methanobacteriales have been identified as natural inhabitants in the human gut. These archaea utilize only methyl compounds including TMA as substrate (Velasquez, Ramezani et al. 2016). Various bacteria grow anaerobically using TMAO as an alternative terminal electron acceptor of a respiratory transport chain (Velasquez, Ramezani et al. 2016). During this energy-yielding reaction, TMAO is reduced to volatile TMA. The bacteria capable of reducing TMAO to TMA are found in different ecological niches such as the marine environment (Photobacterium, Shewanella and Vibrio species), in photosynthetic bacteria living in ponds (Rhodobacter species), and is also found among many enterobacteria (Velasquez, Ramezani et al. 2016). Thus, colonizing human gut with such bacteria could reduce TMAO levels, but this concept needs to be further explored.
Prebiotics
Prebiotics are non-digestible dietary products that can be fermented by the gut microflora and stimulate the growth of beneficial bacteria that colonize the gut (El Hage, Al-Arawe et al. 2023)
Prebiotics and probiotics are both able to modulate the gut microbiome, resulting in favorable effects for the host. A study by Chen et al. discussed the positive effect of resveratrol (RSV), which is a natural polyphenol with prebiotic benefits, on gut health and, specifically, its ability to cause a reduction in TMAO levels in vivo (El Hage, Al-Arawe et al. 2023)
RSV naturally occurs in grapes, berries, and other dietary constituents, and is described to be beneficial in the treatment of many metabolic diseases, including atherosclerosis (El Hage, Al-Arawe et al. 2023); however, its bioavailability is not high. Evidence elucidated that phenolic phytochemicals with poor bioavailability can act through remodeling the gut microbiota(El Hage, Al-Arawe et al. 2023)
For instance, studies found that consumption of RSV can modulate the growth of specific gut microbiota in vivo; this included an increase in the Bacteroidetes-to-Firmicutes ratio, and the growth of Bacteroides, Lactobacillus, and Bifidobacterium (El Hage, Al-Arawe et al. 2023)
Reducing animal protein in the diet
Reduced intake of dietary precursors of TMAO is a potential approach. Dietary l-carnitine has been shown to induce gut microbiota-dependent production of TMA and TMAO in an animal study (Velasquez, Ramezani et al. 2016). . Thus, decreased consumption of l-carnitine and choline could decrease TMAO levels (Velasquez, Ramezani et al. 2016).
Other therapeutics to reduce TMAO
Resveratrol (RSV) is a natural polyphenol with prebiotic benefit that mainly occurs in grapes and berries (Velasquez, Ramezani et al. 2016). Treatment with RSV resulted in an increase in Bacteroidetes abundance at the expense of Firmicutes in choline-treated apoE−/− mice(Velasquez, Ramezani et al. 2016). Serum TMA and TMAO levels were much lower in RSV-treated mice fed with choline than in those in the control group (Velasquez, Ramezani et al. 2016).
Meldonium, an aza-analogue of gamma-butyrobetaine (GBB), competes with quaternary amines, including l-carnitine and GBB, for selected enzymes and transport proteins in the body(Velasquez, Ramezani et al. 2016). . It decreased intestinal microbiota-dependent production of TMA/TMAO from l-carnitine in Wistar rats. Meldonium, however, did not influence bacterial growth and bacterial uptake of l-carnitine.
3-dimethyl-1-butanol (DMB) is a structural analog of choline and an inhibitor for TMA formation through inhibition of microbial TMA lyases. DMB is found in some balsamic vinegar, red wines, and in some olive oils and grape seed oils. DMB inhibited choline diet-enhanced endogenous macrophage foam cell formation and atherosclerotic lesion development in apoE−/− mice without alterations in circulating cholesterol levels (Velasquez, Ramezani et al. 2016). . Furthermore, DMB promoted reduction in microbial taxa that are associated with plasma TMA and TMAO levels and also reduced the choline-diet-dependent enhancement in aortic root atherosclerotic lesion development (Velasquez, Ramezani et al. 2016).
FMO3 is the member of the FMO enzyme family with the highest activity level and is capable of oxidizing TMA to TMAO. A potential site of therapeutic intervention could be at the level of the host enzyme machinery necessary for the conversion of TMA to TMAO. The knockdown of FMO3 using antisense oligonucleotide in mice has been shown to attenuate atherosclerosis. The knockdown of FMO3 not only resulted in decreased TMAO levels but also regulated both lipid metabolism and inflammation, thereby attenuating atherosclerosis. It is important to recall that individuals without functional FMO3 often exhibit a fishy odor, which is caused by trimethylaminuria.
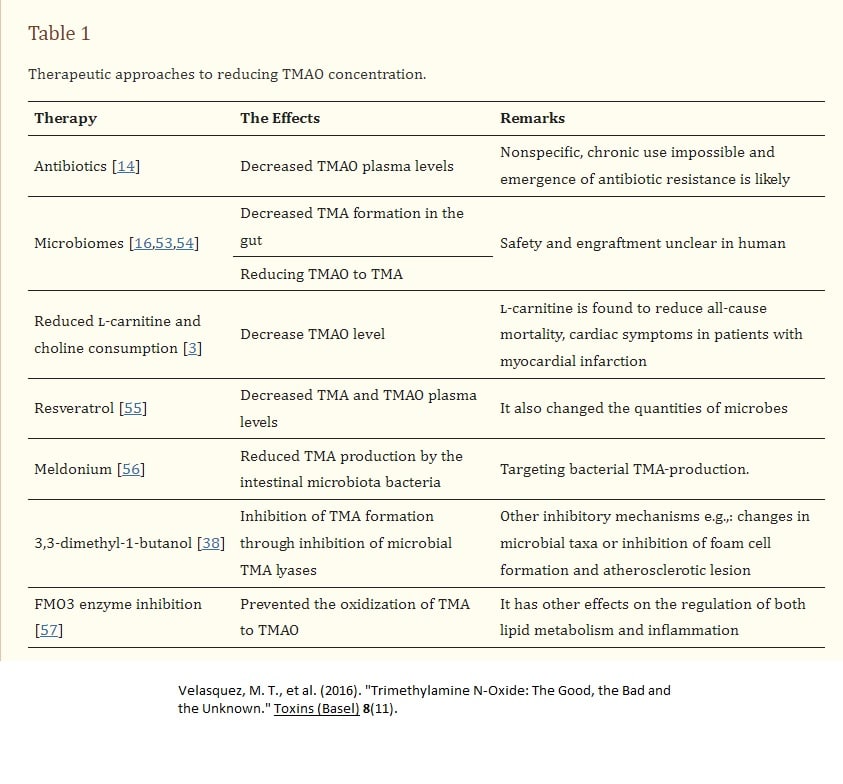
Zeolite
Zeolites are porous minerals with high absorbency and ion-exchange capacity. Their molecular structure is a dense network of AlO4 and SiO4 that generates cavities where water and other polar molecules or ions are inserted/exchanged (Mastinu, Kumar et al. 2019)
Even though there are several synthetic or natural occurring species of zeolites, the most widespread and studied is the naturally occurring zeolite clinoptilolite (ZC) (Mastinu, Kumar et al. 2019)
ZC is an excellent detoxifying, antioxidant and anti-inflammatory agent) (Mastinu, Kumar et al. 2019)
As a result, it is been used in many industrial applications ranging from environmental remediation to oral applications/supplementation in vivo in humans as food supplements or medical devices(Mastinu, Kumar et al. 2019)
.Clinoptilolite is one of the most abundant natural zeolites, widely distributed throughout the world and used for its ion exchange and adsorbent properties (Mastinu, Kumar et al. 2019)
At present, many positive effects are recognized due to the capacity of natural occurring zeolite clinoptilolite (ZC) to adsorb and therefore remove harmful substances like heavy metals, ammonia, or other small molecules in the gastrointestinal tract of humans) (Mastinu, Kumar et al. 2019).
Tribomechanical micronization is a specific micronization first used to activate ZC one time—TMAZ and was further developed as Panaceo Micro Activation technology with PMA-ZC as a result ) (Mastinu, Kumar et al. 2019). Thus obtained, it underwent a very strong ionic excitement, which increased its surface charge and detoxifying capacity towards toxins, radicals, ammonia, and heavy metals) (Mastinu, Kumar et al. 2019).
However, the main element is given by the structural modification making the PMA-ZC much more active in the “recall” in the intestinal lumen) (Mastinu, Kumar et al. 2019). In fact today, PMA-ZC is prescribed for absorbing/chelating and removing harmful and toxic substances from the gastrointestinal tract (e.g., heavy metals, nitrosamines, ammonia, mycotoxins, cations, radioactive materials, pesticides), reducing their absorption in the body) (Mastinu, Kumar et al. 2019).
In addition, a clinical study shows that it strengthens the intestinal wall which might be based on its absorbing properties) (Mastinu, Kumar et al. 2019). Finally, it can also function as an antioxidant by capturing free radicals and reducing the formation of reactive oxygen species, as discussed below) (Mastinu, Kumar et al. 2019).
Most of the clinical positive effects of ZC and modified ZC have been attributed to its reversible ion exchange and adsorption capacity) (Mastinu, Kumar et al. 2019).
In particular, the detoxifying action of PMA-ZC towards ammonia may have potential applications as a therapeutic adjuvant in humans as hypothesized based on results from a clinical study ) (Mastinu, Kumar et al. 2019).Ammonia is produced as a waste in the body during the metabolism of proteins, transformed by the liver into urea, and eliminated by kidneys. Diets rich in proteins, pathologies with excessive protein fermentation, as in the case of irritable bowel and ulcerative colitis lead to an increase in the production of ammonia) (Mastinu, Kumar et al. 2019). High levels of ammonia indicate poor hepatic and renal function. Some authors have highlighted the important ammonia detoxifying contribution of ZC in various diseases ) (Mastinu, Kumar et al. 2019)
Bottom line
Though genetics plays a role in the pathogenesis of IC via some of the coagulation defects mentioned, getting control of inflammation is your first line of defense.
Resources
André, P., et al. (2019). “Metabolic Endotoxemia: A Potential Underlying Mechanism of the Relationship between Dietary Fat Intake and Risk for Cognitive Impairments in Humans?” Nutrients 11(8).
Albagoush SA, Koya S, Chakraborty RK, et al. Factor V Leiden Mutation. [Updated 2023 Apr 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK534802/
Bagameri, L., et al. (2023). “Molecular Insights into Royal Jelly Anti-Inflammatory Properties and Related Diseases.” Life (Basel) 13(7)
Bedran, T. B., et al. (2013). “Fibrinogen-induced streptococcus mutans biofilm formation and adherence to endothelial cells.” Biomed Res Int 2013: 431465.
Cantero, M. A., et al. (2022). “Trimethylamine N-oxide reduction is related to probiotic strain specificity: A systematic review.” Nutr Res 104: 29-35.
Cinelli, M. A., et al. (2020). “Inducible nitric oxide synthase: Regulation, structure, and inhibition.” Med Res Rev 40(1): 158-189.
Den Heijer, M., et al. (2005). “Homocysteine, MTHFR and risk of venous thrombosis: a meta-analysis of published epidemiological studies.” J Thromb Haemost 3(2): 292-299.
Ding, D., et al. (2018). “Further Evidence for Hypercoagulability in Women With Ovarian Endometriomas.” Reprod Sci 25(11): 1540-1548.
Eberly, A. R., et al. (2017). “Biofilm Formation by Uropathogenic Escherichia coli Is Favored under Oxygen Conditions That Mimic the Bladder Environment.” Int J Mol Sci 18(10).
El Hage, R., et al. (2023). “The Role of the Gut Microbiome and Trimethylamine Oxide in Atherosclerosis and Age-Related Disease.” Int J Mol Sci 24(3): 2399.
Gkaliagkousi, E., et al. (2007). “Platelet-Derived Nitric Oxide Signaling and Regulation.” Circulation Research 101(7): 654-662.
Jilani TN, Siddiqui AH. Tissue Plasminogen Activator. [Updated 2023 Feb 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507917/
Kritas, S. K., et al. (2018). “Impact of mold on mast cell-cytokine immune response.” J Biol Regul Homeost Agents 32(4): 763-768.
Levi M, Keller TT, van Gorp E, ten Cate H. Infection and inflammation and the coagulation system. Cardiovasc Res. 2003 Oct 15;60(1):26-39. doi: 10.1016/s0008-6363(02)00857-x. PMID: 14522404.
Lopez MJ, Yandrapalli S. Thrombin. [Updated 2023 May 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557836/
Mannerås-Holm, L., et al. (2011). “Coagulation and Fibrinolytic Disturbances in Women with Polycystic Ovary Syndrome.” The Journal of Clinical Endocrinology & Metabolism 96(4): 1068-1076.
Mastinu, A., et al. (2019). “Zeolite Clinoptilolite: Therapeutic Virtues of an Ancient Mineral.” Molecules 24(8).
McFarlin, B. K., et al. (2017). “Oral spore-based probiotic supplementation was associated with reduced incidence of post-prandial dietary endotoxin, triglycerides, and disease risk biomarkers.” World J Gastrointest Pathophysiol 8(3): 117-126.
Morris, A. A., et al. (2017). “Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency.” J Inherit Metab Dis 40(1): 49-74.
Olas, B. (2022). “The Antioxidant, Anti-Platelet and Anti-Coagulant Properties of Phenolic Compounds, Associated with Modulation of Hemostasis and Cardiovascular Disease, and Their Possible Effect on COVID-19.” Nutrients 14(7).
Qiao, J., et al. (2022). “Trimethylamine N-Oxide Reduces the Susceptibility of Escherichia coli to Multiple Antibiotics.” Front Microbiol 13: 956673.
Seidel, H., et al. (2021). “Effects of Primary Mast Cell Disease on Hemostasis and Erythropoiesis.” Int J Mol Sci 22(16).
Swieringa, F., et al. (2018). “Integrating platelet and coagulation activation in fibrin clot formation.” Res Pract Thromb Haemost 2(3): 450-460.
Valente, M., et al. (2022). “Specialized Pro-Resolving Mediators in Neuroinflammation: Overview of Studies and Perspectives of Clinical Applications.” Molecules 27(15).
Velasquez, M. T., et al. (2016). “Trimethylamine N-Oxide: The Good, the Bad and the Unknown.” Toxins (Basel) 8(11).
Wang, K. Y., et al. (2013). “Recombinant protein production of earthworm lumbrokinase for potential antithrombotic application.” Evid Based Complement Alternat Med 2013: 783971.
You, M. M., et al. (2018). “Royal Jelly Attenuates LPS-Induced Inflammation in BV-2 Microglial Cells through Modulating NF-κB and p38/JNK Signaling Pathways.” Mediators Inflamm 2018: 7834381.
Yu, M., et al. (2021). “The Emerging Role of Mast Cells in Response to Fungal Infection.” Front Immunol 12




")